
Peptide Information
General Information of This Peptide
| Peptide ID |
PEP00161
|
|||||
|---|---|---|---|---|---|---|
| Peptide Name |
Peptide 18-4
|
|||||
| Structure |
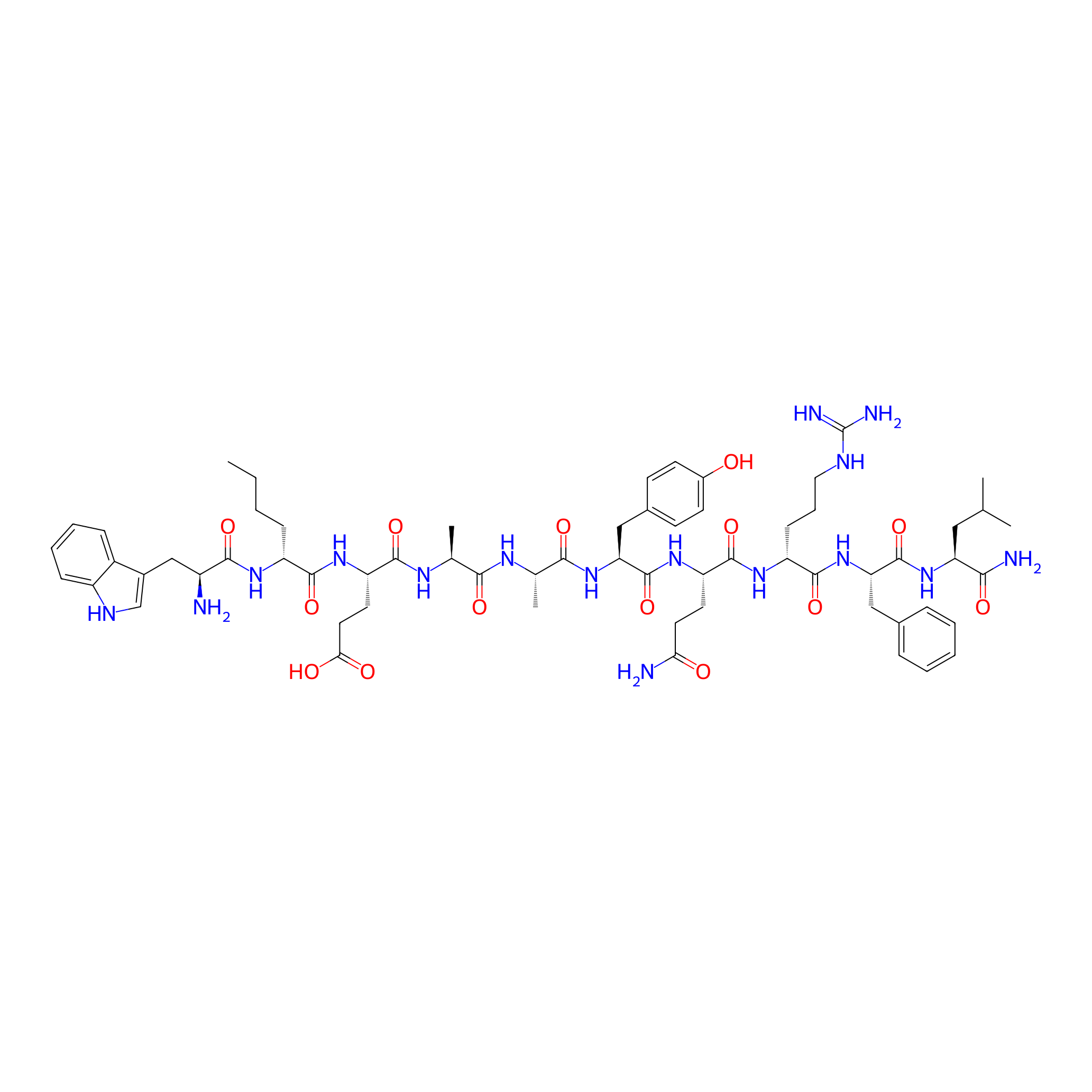
|
|||||
| Sequence |
WXEAAYQrFL
|
|||||
| Peptide Type |
Linear
|
|||||
| Receptor Name |
Keratin, type II cytoskeletal 1 (KRT1)
|
Receptor Info | ||||
| PDC Transmembrane Types | Cell-penetrating peptides (CPPs) | |||||
| Formula |
C63H90N16O14
|
|||||
| Isosmiles |
CCCC[C@@H](NC(=O)[C@@H](N)Cc1c[nH]c2ccccc12)C(=O)N[C@@H](CCC(=O)O)C(=O)N[C@@H](C)C(=O)N[C@@H](C)C(=O)N[C@@H](Cc1ccc(O)cc1)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@H](CCCNC(=N)N)C(=O)N[C@@H](Cc1ccccc1)C(=O)N[C@@H](CC(C)C)C(N)=O
|
|||||
| InChI |
InChI=1S/C63H90N16O14/c1-6-7-17-44(73-56(87)42(64)32-39-33-70-43-18-12-11-16-41(39)43)58(89)75-47(25-27-52(82)83)57(88)72-35(4)54(85)71-36(5)55(86)78-49(31-38-20-22-40(80)23-21-38)61(92)76-46(24-26-51(65)81)60(91)74-45(19-13-28-69-63(67)68)59(90)79-50(30-37-14-9-8-10-15-37)62(93)77-48(53(66)84)29-34(2)3/h8-12,14-16,18,20-23,33-36,42,44-50,70,80H,6-7,13,17,19,24-32,64H2,1-5H3,(H2,65,81)(H2,66,84)(H,71,85)(H,72,88)(H,73,87)(H,74,91)(H,75,89)(H,76,92)(H,77,93)(H,78,86)(H,79,90)(H,82,83)(H4,67,68,69)/t35-,36-,42-,44+,45+,46-,47-,48-,49-,50-/m0/s1
|
|||||
| InChIKey |
LCJLSMZNUNPPOO-YLJKTHCJSA-N
|
|||||
| Pharmaceutical Properties |
Molecule Weight
|
1295.511
|
Polar area
|
509.32
|
||
|
Complexity
|
1294.682242
|
xlogp Value
|
-1.25313
|
|||
|
Heavy Count
|
93
|
Rot Bonds
|
40
|
|||
|
Hbond acc
|
15
|
Hbond Donor
|
18
|
|||
The Activity Data of This Peptide
| Peptide Activity Information 1 | [1] | |||||
| KD | 0.98 μM | |||||
|---|---|---|---|---|---|---|
| Binding Affinity Assay |
A series of SPR experiments were carried out at various concentrations of KRT1 fragment protein (0.1, 1, 10, 100 μM) .
|
|||||
| Experimental Condition | KRT1 fragment protein | |||||
Each Peptide-drug Conjugate Related to This Peptide
Full Information of The Activity Data of The PDC(s) Related to This Peptide
Peptide 18-4 doxorubicin conjugate 1 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
0.9 ± 0.07 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Here we report the design and synthesis of two new peptide-Dox conjugates (1 and 2) for the specific delivery of Dox to the breast cancer cells and the ability to overcome P-glycoprotein multidrug resistance pathway in both drug-sensitive and drug-resistant cancer cells. Peptide-Dox conjugates were evaluated for DOX release in human serum, intracellular delivery compared to free Dox in three cancerous cells (MCF-7, MDA-MB-435, and MDA-MB-435-MDR) and two noncancerous cell lines (HUVEC and MCF-10A), and cytotoxicity compared to free Dox. Results show that both the peptide-Dox conjugates (1 and 2) enter sensitive and resistant cell lines with minimal uptake in normal cells compared to free Dox. Cellular uptake is most likely mediated by a cell specific receptor, as the amount of internalized conjugates significantly decreased in the presence of excess free peptide. Importantly, conjugate 1 is equally cytotoxic as Dox in drug sensitive breast cancer cells and 4 times more potent than free Dox in Dox resistant cell line. Overall, the peptide-Dox ester conjugate 1 showed better breast targeting efficacy than the amide conjugate 2, most likely due to the slow release of Dox from the stable amide linkage.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity experiment was done by incubating the cells with different treatments for 48 h. The results show that conjugate 1 is quite similar to free Dox for toxicity to MCF-7 and MDA-MB-435 cancer cells. In contrast, conjugate 2 was ?20 times less cytotoxic to breast cancerous cells compared to free Dox. This could be attributed to the stability of the amide conjugate 2 inside the cells. Furthermore, the cytotoxicity of conjugate 1 in doxorubicin-resistant cell model MDA-MB-435-MDR (IC50 = 5.4 μM) is 4 times more than free Dox (IC50 = 22 μM). In normal cells (HUVEC and MCF-10A) the two conjugates were 3540 times less toxic compared to breast cancer cells, whereas free Dox was equally cytotoxic (equal IC50) to breast cancer cells and noncancerous cells. Overall these results provide clear evidence that of the two conjugates (conjugate 1 and 2), conjugate 1 has optimal characteristics for specific Dox targeting to breast cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Amelanotic melanoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
1.5 ± 0.09 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Here we report the design and synthesis of two new peptide-Dox conjugates (1 and 2) for the specific delivery of Dox to the breast cancer cells and the ability to overcome P-glycoprotein multidrug resistance pathway in both drug-sensitive and drug-resistant cancer cells. Peptide-Dox conjugates were evaluated for DOX release in human serum, intracellular delivery compared to free Dox in three cancerous cells (MCF-7, MDA-MB-435, and MDA-MB-435-MDR) and two noncancerous cell lines (HUVEC and MCF-10A), and cytotoxicity compared to free Dox. Results show that both the peptide-Dox conjugates (1 and 2) enter sensitive and resistant cell lines with minimal uptake in normal cells compared to free Dox. Cellular uptake is most likely mediated by a cell specific receptor, as the amount of internalized conjugates significantly decreased in the presence of excess free peptide. Importantly, conjugate 1 is equally cytotoxic as Dox in drug sensitive breast cancer cells and 4 times more potent than free Dox in Dox resistant cell line. Overall, the peptide-Dox ester conjugate 1 showed better breast targeting efficacy than the amide conjugate 2, most likely due to the slow release of Dox from the stable amide linkage.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity experiment was done by incubating the cells with different treatments for 48 h. The results show that conjugate 1 is quite similar to free Dox for toxicity to MCF-7 and MDA-MB-435 cancer cells. In contrast, conjugate 2 was ?20 times less cytotoxic to breast cancerous cells compared to free Dox. This could be attributed to the stability of the amide conjugate 2 inside the cells. Furthermore, the cytotoxicity of conjugate 1 in doxorubicin-resistant cell model MDA-MB-435-MDR (IC50 = 5.4 μM) is 4 times more than free Dox (IC50 = 22 μM). In normal cells (HUVEC and MCF-10A) the two conjugates were 3540 times less toxic compared to breast cancer cells, whereas free Dox was equally cytotoxic (equal IC50) to breast cancer cells and noncancerous cells. Overall these results provide clear evidence that of the two conjugates (conjugate 1 and 2), conjugate 1 has optimal characteristics for specific Dox targeting to breast cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Amelanotic melanoma | MDA-MB-435 cell | CVCL_0417 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Amelanotic melanoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
5.4 ± 0.62 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Here we report the design and synthesis of two new peptide-Dox conjugates (1 and 2) for the specific delivery of Dox to the breast cancer cells and the ability to overcome P-glycoprotein multidrug resistance pathway in both drug-sensitive and drug-resistant cancer cells. Peptide-Dox conjugates were evaluated for DOX release in human serum, intracellular delivery compared to free Dox in three cancerous cells (MCF-7, MDA-MB-435, and MDA-MB-435-MDR) and two noncancerous cell lines (HUVEC and MCF-10A), and cytotoxicity compared to free Dox. Results show that both the peptide-Dox conjugates (1 and 2) enter sensitive and resistant cell lines with minimal uptake in normal cells compared to free Dox. Cellular uptake is most likely mediated by a cell specific receptor, as the amount of internalized conjugates significantly decreased in the presence of excess free peptide. Importantly, conjugate 1 is equally cytotoxic as Dox in drug sensitive breast cancer cells and 4 times more potent than free Dox in Dox resistant cell line. Overall, the peptide-Dox ester conjugate 1 showed better breast targeting efficacy than the amide conjugate 2, most likely due to the slow release of Dox from the stable amide linkage.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity experiment was done by incubating the cells with different treatments for 48 h. The results show that conjugate 1 is quite similar to free Dox for toxicity to MCF-7 and MDA-MB-435 cancer cells. In contrast, conjugate 2 was ?20 times less cytotoxic to breast cancerous cells compared to free Dox. This could be attributed to the stability of the amide conjugate 2 inside the cells. Furthermore, the cytotoxicity of conjugate 1 in doxorubicin-resistant cell model MDA-MB-435-MDR (IC50 = 5.4 μM) is 4 times more than free Dox (IC50 = 22 μM). In normal cells (HUVEC and MCF-10A) the two conjugates were 3540 times less toxic compared to breast cancer cells, whereas free Dox was equally cytotoxic (equal IC50) to breast cancer cells and noncancerous cells. Overall these results provide clear evidence that of the two conjugates (conjugate 1 and 2), conjugate 1 has optimal characteristics for specific Dox targeting to breast cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Amelanotic melanoma | MDA-MB-435 cell | CVCL_0417 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
35.1 ± 2.2 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Here we report the design and synthesis of two new peptide-Dox conjugates (1 and 2) for the specific delivery of Dox to the breast cancer cells and the ability to overcome P-glycoprotein multidrug resistance pathway in both drug-sensitive and drug-resistant cancer cells. Peptide-Dox conjugates were evaluated for DOX release in human serum, intracellular delivery compared to free Dox in three cancerous cells (MCF-7, MDA-MB-435, and MDA-MB-435-MDR) and two noncancerous cell lines (HUVEC and MCF-10A), and cytotoxicity compared to free Dox. Results show that both the peptide-Dox conjugates (1 and 2) enter sensitive and resistant cell lines with minimal uptake in normal cells compared to free Dox. Cellular uptake is most likely mediated by a cell specific receptor, as the amount of internalized conjugates significantly decreased in the presence of excess free peptide. Importantly, conjugate 1 is equally cytotoxic as Dox in drug sensitive breast cancer cells and 4 times more potent than free Dox in Dox resistant cell line. Overall, the peptide-Dox ester conjugate 1 showed better breast targeting efficacy than the amide conjugate 2, most likely due to the slow release of Dox from the stable amide linkage.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity experiment was done by incubating the cells with different treatments for 48 h. The results show that conjugate 1 is quite similar to free Dox for toxicity to MCF-7 and MDA-MB-435 cancer cells. In contrast, conjugate 2 was ?20 times less cytotoxic to breast cancerous cells compared to free Dox. This could be attributed to the stability of the amide conjugate 2 inside the cells. Furthermore, the cytotoxicity of conjugate 1 in doxorubicin-resistant cell model MDA-MB-435-MDR (IC50 = 5.4 μM) is 4 times more than free Dox (IC50 = 22 μM). In normal cells (HUVEC and MCF-10A) the two conjugates were 3540 times less toxic compared to breast cancer cells, whereas free Dox was equally cytotoxic (equal IC50) to breast cancer cells and noncancerous cells. Overall these results provide clear evidence that of the two conjugates (conjugate 1 and 2), conjugate 1 has optimal characteristics for specific Dox targeting to breast cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
42.3 ± 2.4 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Here we report the design and synthesis of two new peptide-Dox conjugates (1 and 2) for the specific delivery of Dox to the breast cancer cells and the ability to overcome P-glycoprotein multidrug resistance pathway in both drug-sensitive and drug-resistant cancer cells. Peptide-Dox conjugates were evaluated for DOX release in human serum, intracellular delivery compared to free Dox in three cancerous cells (MCF-7, MDA-MB-435, and MDA-MB-435-MDR) and two noncancerous cell lines (HUVEC and MCF-10A), and cytotoxicity compared to free Dox. Results show that both the peptide-Dox conjugates (1 and 2) enter sensitive and resistant cell lines with minimal uptake in normal cells compared to free Dox. Cellular uptake is most likely mediated by a cell specific receptor, as the amount of internalized conjugates significantly decreased in the presence of excess free peptide. Importantly, conjugate 1 is equally cytotoxic as Dox in drug sensitive breast cancer cells and 4 times more potent than free Dox in Dox resistant cell line. Overall, the peptide-Dox ester conjugate 1 showed better breast targeting efficacy than the amide conjugate 2, most likely due to the slow release of Dox from the stable amide linkage.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity experiment was done by incubating the cells with different treatments for 48 h. The results show that conjugate 1 is quite similar to free Dox for toxicity to MCF-7 and MDA-MB-435 cancer cells. In contrast, conjugate 2 was ?20 times less cytotoxic to breast cancerous cells compared to free Dox. This could be attributed to the stability of the amide conjugate 2 inside the cells. Furthermore, the cytotoxicity of conjugate 1 in doxorubicin-resistant cell model MDA-MB-435-MDR (IC50 = 5.4 μM) is 4 times more than free Dox (IC50 = 22 μM). In normal cells (HUVEC and MCF-10A) the two conjugates were 3540 times less toxic compared to breast cancer cells, whereas free Dox was equally cytotoxic (equal IC50) to breast cancer cells and noncancerous cells. Overall these results provide clear evidence that of the two conjugates (conjugate 1 and 2), conjugate 1 has optimal characteristics for specific Dox targeting to breast cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Human umbilical vein endothelial cell | Homo sapiens | ||
Peptide 18-4-doxorubicin [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
1.3 µM
|
|||
| Evaluation Method | MTT assay | ||||
| Description |
The results showed that the cytotoxicity of both conjugates 1 and 2 (IC50 = 1.3 and 2.2 uM, respectively), as well as the free Dox (IC50 = 1.5 uM) on MDA-MB-231 breast cancer cell line, were in the low micromolar range (Figure 4). For the breast cancer cell line MDA-MB-468, the free Dox (IC50 = 0.35 uM) was slightly more toxic compared to conjugates 1 (4.7 uM) and 2 (1.2 uM). For the non-cancerous cell line MCF 10A, the free Dox was highly toxic (IC50 = 0.24 uM) whereas conjugates 1 and 2 displayed much-reduced toxicity (IC50 = 38.6 and 15.1 uM, respectively).
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
1.3 ± 0.2µM
|
|||
| MOA of PDC |
We engineered peptides that bind to cell-surface K1 and are internalized by breast cancer cells via cell-surface K1 receptor-mediated endocytosis. Peptide 18-4 (WxEAAYQrFL), with two D-amino acids, is a second-generation breast cancer cell-targeting peptide that is proteolytically stable (100% intact up to 24 h when incubated with human serum or liver homogenate from mice) and has shown high specific uptake by breast cancer cells and minimal/no binding to non-cancerous cells. Affinity purification of breast cancer cell lysates using the immobilized peptide, followed by liquid chromatography-tandem mass spectrometry and proteomics were used to identify K1 as the novel target for peptide 18-4 in cancer cells. Further, we showed that the uptake of the peptide by the cancer cells is dependent on K1 expression.
Click to Show/Hide
|
||||
| Description |
Several PDCs have been prepared with peptide 18-4 and doxorubicin (Dox) using different linker chemistries such as ester, amide, succinimidyl thioether, or acyl hydrazone. In vitro results showed that these PDCs were highly specific toward breast cancer cells. PDCs displayed similar toxicity as free Dox toward the breast cancer cells and several-fold (7-40 times) less toxicity toward the non-cancerous cells such as MCF-10A and human umbilical vein endothelial cells (HUVECs). A peptide 18-4-Dox conjugate with amide/succinimidyl thioether linkage showed high selective toxicity toward triple negative breast cancer (TNBC) cell lines, i.e., MDA-MB-231 cells (IC50 1.3 ± 0.2 uM) and MDA-MB-468 cells (IC50 4.7 ± 0.3 uM) compared to the normal breast MCF10A cells (IC50 38.6 ± 1.1 uM). The linkage between the drug and the peptide was stable as the degradation half-life of peptide 18-4-Dox conjugate in the presence of human serum was found to be ˜18 h. Herein, we describe the first in vivo evidence for improved efficacy of this PDC targeting K1 receptor in an orthotopic TNBC mouse model. We also show a higher accumulation of PDC in TNBC tumors in mice, in accord with K1 overexpression in tumor over non-tumor tissues in MDA-MB-231 xenografted mice.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
4.7 µM
|
|||
| Evaluation Method | MTT assay | ||||
| Description |
The results showed that the cytotoxicity of both conjugates 1 and 2 (IC50 = 1.3 and 2.2 uM, respectively), as well as the free Dox (IC50 = 1.5 uM) on MDA-MB-231 breast cancer cell line, were in the low micromolar range (Figure 4). For the breast cancer cell line MDA-MB-468, the free Dox (IC50 = 0.35 uM) was slightly more toxic compared to conjugates 1 (4.7 uM) and 2 (1.2 uM). For the non-cancerous cell line MCF 10A, the free Dox was highly toxic (IC50 = 0.24 uM) whereas conjugates 1 and 2 displayed much-reduced toxicity (IC50 = 38.6 and 15.1 uM, respectively).
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-468 cell | CVCL_0419 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
4.7 ± 0.3µM
|
|||
| MOA of PDC |
We engineered peptides that bind to cell-surface K1 and are internalized by breast cancer cells via cell-surface K1 receptor-mediated endocytosis. Peptide 18-4 (WxEAAYQrFL), with two D-amino acids, is a second-generation breast cancer cell-targeting peptide that is proteolytically stable (100% intact up to 24 h when incubated with human serum or liver homogenate from mice) and has shown high specific uptake by breast cancer cells and minimal/no binding to non-cancerous cells. Affinity purification of breast cancer cell lysates using the immobilized peptide, followed by liquid chromatography-tandem mass spectrometry and proteomics were used to identify K1 as the novel target for peptide 18-4 in cancer cells. Further, we showed that the uptake of the peptide by the cancer cells is dependent on K1 expression.
Click to Show/Hide
|
||||
| Description |
Several PDCs have been prepared with peptide 18-4 and doxorubicin (Dox) using different linker chemistries such as ester, amide, succinimidyl thioether, or acyl hydrazone. In vitro results showed that these PDCs were highly specific toward breast cancer cells. PDCs displayed similar toxicity as free Dox toward the breast cancer cells and several-fold (7-40 times) less toxicity toward the non-cancerous cells such as MCF-10A and human umbilical vein endothelial cells (HUVECs). A peptide 18-4-Dox conjugate with amide/succinimidyl thioether linkage showed high selective toxicity toward triple negative breast cancer (TNBC) cell lines, i.e., MDA-MB-231 cells (IC50 1.3 ± 0.2 uM) and MDA-MB-468 cells (IC50 4.7 ± 0.3 uM) compared to the normal breast MCF10A cells (IC50 38.6 ± 1.1 uM). The linkage between the drug and the peptide was stable as the degradation half-life of peptide 18-4-Dox conjugate in the presence of human serum was found to be ˜18 h. Herein, we describe the first in vivo evidence for improved efficacy of this PDC targeting K1 receptor in an orthotopic TNBC mouse model. We also show a higher accumulation of PDC in TNBC tumors in mice, in accord with K1 overexpression in tumor over non-tumor tissues in MDA-MB-231 xenografted mice.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-468 cell | CVCL_0419 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
38.6 µM
|
|||
| Evaluation Method | MTT assay | ||||
| Description |
The results showed that the cytotoxicity of both conjugates 1 and 2 (IC50 = 1.3 and 2.2 uM, respectively), as well as the free Dox (IC50 = 1.5 uM) on MDA-MB-231 breast cancer cell line, were in the low micromolar range (Figure 4). For the breast cancer cell line MDA-MB-468, the free Dox (IC50 = 0.35 uM) was slightly more toxic compared to conjugates 1 (4.7 uM) and 2 (1.2 uM). For the non-cancerous cell line MCF 10A, the free Dox was highly toxic (IC50 = 0.24 uM) whereas conjugates 1 and 2 displayed much-reduced toxicity (IC50 = 38.6 and 15.1 uM, respectively).
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
38.6 ± 1.1µM
|
|||
| MOA of PDC |
We engineered peptides that bind to cell-surface K1 and are internalized by breast cancer cells via cell-surface K1 receptor-mediated endocytosis. Peptide 18-4 (WxEAAYQrFL), with two D-amino acids, is a second-generation breast cancer cell-targeting peptide that is proteolytically stable (100% intact up to 24 h when incubated with human serum or liver homogenate from mice) and has shown high specific uptake by breast cancer cells and minimal/no binding to non-cancerous cells. Affinity purification of breast cancer cell lysates using the immobilized peptide, followed by liquid chromatography-tandem mass spectrometry and proteomics were used to identify K1 as the novel target for peptide 18-4 in cancer cells. Further, we showed that the uptake of the peptide by the cancer cells is dependent on K1 expression.
Click to Show/Hide
|
||||
| Description |
Several PDCs have been prepared with peptide 18-4 and doxorubicin (Dox) using different linker chemistries such as ester, amide, succinimidyl thioether, or acyl hydrazone. In vitro results showed that these PDCs were highly specific toward breast cancer cells. PDCs displayed similar toxicity as free Dox toward the breast cancer cells and several-fold (7-40 times) less toxicity toward the non-cancerous cells such as MCF-10A and human umbilical vein endothelial cells (HUVECs). A peptide 18-4-Dox conjugate with amide/succinimidyl thioether linkage showed high selective toxicity toward triple negative breast cancer (TNBC) cell lines, i.e., MDA-MB-231 cells (IC50 1.3 ± 0.2 uM) and MDA-MB-468 cells (IC50 4.7 ± 0.3 uM) compared to the normal breast MCF10A cells (IC50 38.6 ± 1.1 uM). The linkage between the drug and the peptide was stable as the degradation half-life of peptide 18-4-Dox conjugate in the presence of human serum was found to be ˜18 h. Herein, we describe the first in vivo evidence for improved efficacy of this PDC targeting K1 receptor in an orthotopic TNBC mouse model. We also show a higher accumulation of PDC in TNBC tumors in mice, in accord with K1 overexpression in tumor over non-tumor tissues in MDA-MB-231 xenografted mice.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
Peptide 18-4 doxorubicin conjugate 2 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Amelanotic melanoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
18.6 ± 2.5 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Here we report the design and synthesis of two new peptide-Dox conjugates (1 and 2) for the specific delivery of Dox to the breast cancer cells and the ability to overcome P-glycoprotein multidrug resistance pathway in both drug-sensitive and drug-resistant cancer cells. Peptide-Dox conjugates were evaluated for DOX release in human serum, intracellular delivery compared to free Dox in three cancerous cells (MCF-7, MDA-MB-435, and MDA-MB-435-MDR) and two noncancerous cell lines (HUVEC and MCF-10A), and cytotoxicity compared to free Dox. Results show that both the peptide-Dox conjugates (1 and 2) enter sensitive and resistant cell lines with minimal uptake in normal cells compared to free Dox. Cellular uptake is most likely mediated by a cell specific receptor, as the amount of internalized conjugates significantly decreased in the presence of excess free peptide. Importantly, conjugate 1 is equally cytotoxic as Dox in drug sensitive breast cancer cells and 4 times more potent than free Dox in Dox resistant cell line. Overall, the peptide-Dox ester conjugate 1 showed better breast targeting efficacy than the amide conjugate 2, most likely due to the slow release of Dox from the stable amide linkage.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity experiment was done by incubating the cells with different treatments for 48 h. The results show that conjugate 1 is quite similar to free Dox for toxicity to MCF-7 and MDA-MB-435 cancer cells. In contrast, conjugate 2 was ?20 times less cytotoxic to breast cancerous cells compared to free Dox. This could be attributed to the stability of the amide conjugate 2 inside the cells. Furthermore, the cytotoxicity of conjugate 1 in doxorubicin-resistant cell model MDA-MB-435-MDR (IC50 = 5.4 μM) is 4 times more than free Dox (IC50 = 22 μM). In normal cells (HUVEC and MCF-10A) the two conjugates were 3540 times less toxic compared to breast cancer cells, whereas free Dox was equally cytotoxic (equal IC50) to breast cancer cells and noncancerous cells. Overall these results provide clear evidence that of the two conjugates (conjugate 1 and 2), conjugate 1 has optimal characteristics for specific Dox targeting to breast cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Amelanotic melanoma | MDA-MB-435 cell | CVCL_0417 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Amelanotic melanoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
19.7 ± 3.1 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Here we report the design and synthesis of two new peptide-Dox conjugates (1 and 2) for the specific delivery of Dox to the breast cancer cells and the ability to overcome P-glycoprotein multidrug resistance pathway in both drug-sensitive and drug-resistant cancer cells. Peptide-Dox conjugates were evaluated for DOX release in human serum, intracellular delivery compared to free Dox in three cancerous cells (MCF-7, MDA-MB-435, and MDA-MB-435-MDR) and two noncancerous cell lines (HUVEC and MCF-10A), and cytotoxicity compared to free Dox. Results show that both the peptide-Dox conjugates (1 and 2) enter sensitive and resistant cell lines with minimal uptake in normal cells compared to free Dox. Cellular uptake is most likely mediated by a cell specific receptor, as the amount of internalized conjugates significantly decreased in the presence of excess free peptide. Importantly, conjugate 1 is equally cytotoxic as Dox in drug sensitive breast cancer cells and 4 times more potent than free Dox in Dox resistant cell line. Overall, the peptide-Dox ester conjugate 1 showed better breast targeting efficacy than the amide conjugate 2, most likely due to the slow release of Dox from the stable amide linkage.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity experiment was done by incubating the cells with different treatments for 48 h. The results show that conjugate 1 is quite similar to free Dox for toxicity to MCF-7 and MDA-MB-435 cancer cells. In contrast, conjugate 2 was ?20 times less cytotoxic to breast cancerous cells compared to free Dox. This could be attributed to the stability of the amide conjugate 2 inside the cells. Furthermore, the cytotoxicity of conjugate 1 in doxorubicin-resistant cell model MDA-MB-435-MDR (IC50 = 5.4 μM) is 4 times more than free Dox (IC50 = 22 μM). In normal cells (HUVEC and MCF-10A) the two conjugates were 3540 times less toxic compared to breast cancer cells, whereas free Dox was equally cytotoxic (equal IC50) to breast cancer cells and noncancerous cells. Overall these results provide clear evidence that of the two conjugates (conjugate 1 and 2), conjugate 1 has optimal characteristics for specific Dox targeting to breast cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Amelanotic melanoma | MDA-MB-435 cell | CVCL_0417 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
40.5 ± 4.3 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Here we report the design and synthesis of two new peptide-Dox conjugates (1 and 2) for the specific delivery of Dox to the breast cancer cells and the ability to overcome P-glycoprotein multidrug resistance pathway in both drug-sensitive and drug-resistant cancer cells. Peptide-Dox conjugates were evaluated for DOX release in human serum, intracellular delivery compared to free Dox in three cancerous cells (MCF-7, MDA-MB-435, and MDA-MB-435-MDR) and two noncancerous cell lines (HUVEC and MCF-10A), and cytotoxicity compared to free Dox. Results show that both the peptide-Dox conjugates (1 and 2) enter sensitive and resistant cell lines with minimal uptake in normal cells compared to free Dox. Cellular uptake is most likely mediated by a cell specific receptor, as the amount of internalized conjugates significantly decreased in the presence of excess free peptide. Importantly, conjugate 1 is equally cytotoxic as Dox in drug sensitive breast cancer cells and 4 times more potent than free Dox in Dox resistant cell line. Overall, the peptide-Dox ester conjugate 1 showed better breast targeting efficacy than the amide conjugate 2, most likely due to the slow release of Dox from the stable amide linkage.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity experiment was done by incubating the cells with different treatments for 48 h. The results show that conjugate 1 is quite similar to free Dox for toxicity to MCF-7 and MDA-MB-435 cancer cells. In contrast, conjugate 2 was ?20 times less cytotoxic to breast cancerous cells compared to free Dox. This could be attributed to the stability of the amide conjugate 2 inside the cells. Furthermore, the cytotoxicity of conjugate 1 in doxorubicin-resistant cell model MDA-MB-435-MDR (IC50 = 5.4 μM) is 4 times more than free Dox (IC50 = 22 μM). In normal cells (HUVEC and MCF-10A) the two conjugates were 3540 times less toxic compared to breast cancer cells, whereas free Dox was equally cytotoxic (equal IC50) to breast cancer cells and noncancerous cells. Overall these results provide clear evidence that of the two conjugates (conjugate 1 and 2), conjugate 1 has optimal characteristics for specific Dox targeting to breast cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
50.9 ± 3.2 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Here we report the design and synthesis of two new peptide-Dox conjugates (1 and 2) for the specific delivery of Dox to the breast cancer cells and the ability to overcome P-glycoprotein multidrug resistance pathway in both drug-sensitive and drug-resistant cancer cells. Peptide-Dox conjugates were evaluated for DOX release in human serum, intracellular delivery compared to free Dox in three cancerous cells (MCF-7, MDA-MB-435, and MDA-MB-435-MDR) and two noncancerous cell lines (HUVEC and MCF-10A), and cytotoxicity compared to free Dox. Results show that both the peptide-Dox conjugates (1 and 2) enter sensitive and resistant cell lines with minimal uptake in normal cells compared to free Dox. Cellular uptake is most likely mediated by a cell specific receptor, as the amount of internalized conjugates significantly decreased in the presence of excess free peptide. Importantly, conjugate 1 is equally cytotoxic as Dox in drug sensitive breast cancer cells and 4 times more potent than free Dox in Dox resistant cell line. Overall, the peptide-Dox ester conjugate 1 showed better breast targeting efficacy than the amide conjugate 2, most likely due to the slow release of Dox from the stable amide linkage.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity experiment was done by incubating the cells with different treatments for 48 h. The results show that conjugate 1 is quite similar to free Dox for toxicity to MCF-7 and MDA-MB-435 cancer cells. In contrast, conjugate 2 was ?20 times less cytotoxic to breast cancerous cells compared to free Dox. This could be attributed to the stability of the amide conjugate 2 inside the cells. Furthermore, the cytotoxicity of conjugate 1 in doxorubicin-resistant cell model MDA-MB-435-MDR (IC50 = 5.4 μM) is 4 times more than free Dox (IC50 = 22 μM). In normal cells (HUVEC and MCF-10A) the two conjugates were 3540 times less toxic compared to breast cancer cells, whereas free Dox was equally cytotoxic (equal IC50) to breast cancer cells and noncancerous cells. Overall these results provide clear evidence that of the two conjugates (conjugate 1 and 2), conjugate 1 has optimal characteristics for specific Dox targeting to breast cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Human umbilical vein endothelial cell | Homo sapiens | ||
| Experiment 5 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
191 ± 2.8 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Here we report the design and synthesis of two new peptide-Dox conjugates (1 and 2) for the specific delivery of Dox to the breast cancer cells and the ability to overcome P-glycoprotein multidrug resistance pathway in both drug-sensitive and drug-resistant cancer cells. Peptide-Dox conjugates were evaluated for DOX release in human serum, intracellular delivery compared to free Dox in three cancerous cells (MCF-7, MDA-MB-435, and MDA-MB-435-MDR) and two noncancerous cell lines (HUVEC and MCF-10A), and cytotoxicity compared to free Dox. Results show that both the peptide-Dox conjugates (1 and 2) enter sensitive and resistant cell lines with minimal uptake in normal cells compared to free Dox. Cellular uptake is most likely mediated by a cell specific receptor, as the amount of internalized conjugates significantly decreased in the presence of excess free peptide. Importantly, conjugate 1 is equally cytotoxic as Dox in drug sensitive breast cancer cells and 4 times more potent than free Dox in Dox resistant cell line. Overall, the peptide-Dox ester conjugate 1 showed better breast targeting efficacy than the amide conjugate 2, most likely due to the slow release of Dox from the stable amide linkage.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity experiment was done by incubating the cells with different treatments for 48 h. The results show that conjugate 1 is quite similar to free Dox for toxicity to MCF-7 and MDA-MB-435 cancer cells. In contrast, conjugate 2 was ?20 times less cytotoxic to breast cancerous cells compared to free Dox. This could be attributed to the stability of the amide conjugate 2 inside the cells. Furthermore, the cytotoxicity of conjugate 1 in doxorubicin-resistant cell model MDA-MB-435-MDR (IC50 = 5.4 μM) is 4 times more than free Dox (IC50 = 22 μM). In normal cells (HUVEC and MCF-10A) the two conjugates were 3540 times less toxic compared to breast cancer cells, whereas free Dox was equally cytotoxic (equal IC50) to breast cancer cells and noncancerous cells. Overall these results provide clear evidence that of the two conjugates (conjugate 1 and 2), conjugate 1 has optimal characteristics for specific Dox targeting to breast cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
POSS-p18-4-Ce6(PPC) [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Cell viability |
10%
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Polyhedral oligomeric silsesquioxane (POSS) molecules have a distinct nanostructure, consisting of an inner inorganic cage core of silicon and oxygen atoms and an outer shell of organic functional groups. The unique structure of POSS molecules containing reactive functionalities and their superior biocompatibility make them suitable drug-delivery carriers. In the present study, we prepared p 18-4/chlorin e6 (Ce6)-conjugated POSS (PPC) nanoparticles for improving the targeting ability of Ce6 to breast cancer cells. To fabricate PPC nanoparticles, p 18-4 was first covalently conjugated on OctaMaleamicacid POSS (OM-POSS), and subsequently, amino-terminated Ce6 was introduced. To verify the availability of the PPC nanoparticles for cancer therapy, their physicochemical properties, including morphology, chemical structure, and particle size, were systematically characterized. The cellular uptake, phototoxicity, and targeting ability of the PPC nanoparticles were assessed using a human breast cancer cell line MDA-MB-231. In addition, PPC nanoparticles was injected intravenously into tumor-bearing mice to evaluate in vivo biodistribution and PDT efficacy.
Click to Show/Hide
|
||||
| Description |
The internalization of nanoparticles in cancer cells is the foremost step in PDT. Targeting ligand decoration and reducing nanoparticle size have been intensively explored to enhance the intracellular uptake of nanoparticles. Therefore, the PPC nanoparticles were designed to offer the appropriate size and targeting ability for delivery of Ce6 to tumor tissues via active and passive targeting mechanisms. The cellular internalization of free Ce6 and PPC into various cancer cells, i.e., MDA-MB-231, MKN-28, and HeLa, was assessed by fluorescence-activated cell sorting (FACS) analysis using a flow cytometer. When compared with free Ce6, the higher cellular internalization of PPC was observed in all cancer cells tested. In particular, the fluorescence intensity due to the intracellular uptake of PPC in MDA-MB-231 cells increased more dramatically than in the other cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
References