
Linker Information
General Information of This Linker
| Linker ID |
LIN00033
|
|||||
|---|---|---|---|---|---|---|
| Linker Name |
Glutaric acid
|
|||||
| Structure |
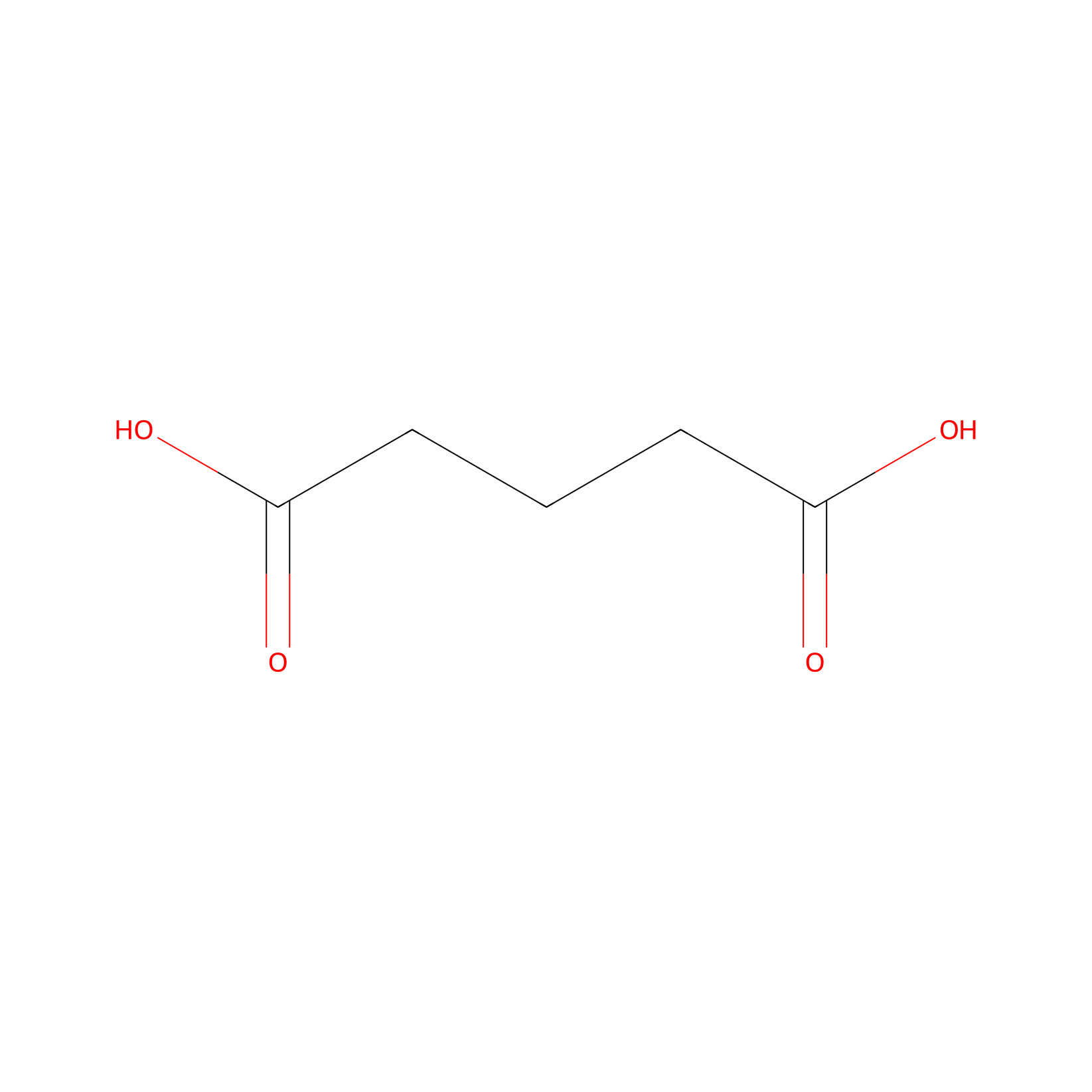
|
|||||
| Formula |
C5H8O4
|
|||||
| #Ro5 Violations (Lipinski): 0 | Molecular Weight (mw) | 132.11 | ||||
| Lipid-water partition coefficient (xlogp) | -0.3 | |||||
| Hydrogen Bond Donor Count (hbonddonor) | 2 | |||||
| Hydrogen Bond Acceptor Count (hbondacc) | 4 | |||||
| Rotatable Bond Count (rotbonds) | 4 | |||||
| Chemble ID | ||||||
| Chemble ID | ||||||
| PubChem CID | ||||||
| Canonical smiles |
C(CC(=O)O)CC(=O)O
|
|||||
| InChI |
InChI=1S/C5H8O4/c6-4(7)2-1-3-5(8)9/h1-3H2,(H,6,7)(H,8,9)
|
|||||
| InChIKey |
JFCQEDHGNNZCLN-UHFFFAOYSA-N
|
|||||
| IUPAC Name |
pentanedioic acid
|
|||||
Each Peptide-drug Conjugate Related to This Linker
Full Information of The Activity Data of The PDC(s) Related to This Linker
R-C12-4 [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
38.00%
|
|||
| Administration Time | 6 days | ||||
| Administration Dosage | 200 μL 15 mg/kg | ||||
| MOA of PDC |
In this study, we would investigate the co-modification of fatty acids and monosaccharides in anticancer peptides, as we expect to optimize the selectivity and cytotoxicity of the peptide at the same time. Based on our previous study, the lipopeptide R-C12 was selected as a template to perform glucose derivative modification at different positions as we supposed that the glucose derivative modification position in R-C12 might be a crucial factor in its selectivity and activity optimization. The glucose derivative was covalently coupled to different sites in R-C12 through its -amino group of a Lys residue, which was obtained by replacing the Arg residue. R-C12 contains seven Arg residues, accordingly, seven glycolipid peptides were designed and synthesized, which were named R-C12-1, R-C12-2, R-C12-3, R-C12-4, R-C12-5, R-C12-6, and R-C12-7. Various physiochemical properties including the hydrodynamic size, ζ-potential, secondary structure, and hydrophobicity were determined to explain the different cytotoxicity and selectivity of these glycolipid peptides. On the other hand, the strength of the binding between glycolipid peptides and the cancer cells mediated by GLUT1 has also been explored. The most optimized glycolipid peptide R-C12-4 demonstrated significant cytotoxicity and antimetastasis in vitro and in vivo, which indicated that it may be a good lead for anticancer drug development.
Click to Show/Hide
|
||||
| Description |
For the subcutaneous transplantation model, R-C12 or R-C12-4 was injected intraperitoneally every other day at the dose of 15 mg/kg. As shown in Figure 7A,B, after 12 days, R-C12-4 and R-C12 have a significant inhibition effect on the BALB/c mice tumor volume compared to the control group (p < 0.01 and p < 0.05, respectively). For the metastasis model, R-C12 or R-C12-4 was applied by the tail intravenous injection every 2 days at the dose of 10 mg/kg. As shown in Figure 7D, significantly fewer metastatic nodules from the lungs of Kunming (KM) mice were observed in the R-C12-4 and R-C12 treatment groups compared to the control and this result was confirmed by the H&E staining of lung sections. Visible tumor nodules were counted on the surface of the lung demonstrating a significant difference between the control and R-C12 and R-C12-4 treatment groups (p < 0.05). It is noteworthy that both R-C12 and R-C12-4 did not affect the mouse body weight either in the subcutaneous transplantation model or in the metastasis model, indicating that these peptides did not diminish their overall health.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c mice B16-F10 cells xenograft model. | ||||
| In Vitro Model | Mouse melanoma | B16-F10 cell | CVCL_0159 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
50.00%
|
|||
| Administration Time | 3 days | ||||
| Administration Dosage | 200 μL 15 mg/kg | ||||
| MOA of PDC |
In this study, we would investigate the co-modification of fatty acids and monosaccharides in anticancer peptides, as we expect to optimize the selectivity and cytotoxicity of the peptide at the same time. Based on our previous study, the lipopeptide R-C12 was selected as a template to perform glucose derivative modification at different positions as we supposed that the glucose derivative modification position in R-C12 might be a crucial factor in its selectivity and activity optimization. The glucose derivative was covalently coupled to different sites in R-C12 through its -amino group of a Lys residue, which was obtained by replacing the Arg residue. R-C12 contains seven Arg residues, accordingly, seven glycolipid peptides were designed and synthesized, which were named R-C12-1, R-C12-2, R-C12-3, R-C12-4, R-C12-5, R-C12-6, and R-C12-7. Various physiochemical properties including the hydrodynamic size, ζ-potential, secondary structure, and hydrophobicity were determined to explain the different cytotoxicity and selectivity of these glycolipid peptides. On the other hand, the strength of the binding between glycolipid peptides and the cancer cells mediated by GLUT1 has also been explored. The most optimized glycolipid peptide R-C12-4 demonstrated significant cytotoxicity and antimetastasis in vitro and in vivo, which indicated that it may be a good lead for anticancer drug development.
Click to Show/Hide
|
||||
| Description |
For the subcutaneous transplantation model, R-C12 or R-C12-4 was injected intraperitoneally every other day at the dose of 15 mg/kg. As shown in Figure 7A,B, after 12 days, R-C12-4 and R-C12 have a significant inhibition effect on the BALB/c mice tumor volume compared to the control group (p < 0.01 and p < 0.05, respectively). For the metastasis model, R-C12 or R-C12-4 was applied by the tail intravenous injection every 2 days at the dose of 10 mg/kg. As shown in Figure 7D, significantly fewer metastatic nodules from the lungs of Kunming (KM) mice were observed in the R-C12-4 and R-C12 treatment groups compared to the control and this result was confirmed by the H&E staining of lung sections. Visible tumor nodules were counted on the surface of the lung demonstrating a significant difference between the control and R-C12 and R-C12-4 treatment groups (p < 0.05). It is noteworthy that both R-C12 and R-C12-4 did not affect the mouse body weight either in the subcutaneous transplantation model or in the metastasis model, indicating that these peptides did not diminish their overall health.
Click to Show/Hide
|
||||
| In Vivo Model | KM mice B16-F10 cells xenograft model. | ||||
| In Vitro Model | Mouse melanoma | B16-F10 cell | CVCL_0159 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
62.50%
|
|||
| Administration Time | 9 days | ||||
| Administration Dosage | 200 μL 15 mg/kg | ||||
| MOA of PDC |
In this study, we would investigate the co-modification of fatty acids and monosaccharides in anticancer peptides, as we expect to optimize the selectivity and cytotoxicity of the peptide at the same time. Based on our previous study, the lipopeptide R-C12 was selected as a template to perform glucose derivative modification at different positions as we supposed that the glucose derivative modification position in R-C12 might be a crucial factor in its selectivity and activity optimization. The glucose derivative was covalently coupled to different sites in R-C12 through its -amino group of a Lys residue, which was obtained by replacing the Arg residue. R-C12 contains seven Arg residues, accordingly, seven glycolipid peptides were designed and synthesized, which were named R-C12-1, R-C12-2, R-C12-3, R-C12-4, R-C12-5, R-C12-6, and R-C12-7. Various physiochemical properties including the hydrodynamic size, ζ-potential, secondary structure, and hydrophobicity were determined to explain the different cytotoxicity and selectivity of these glycolipid peptides. On the other hand, the strength of the binding between glycolipid peptides and the cancer cells mediated by GLUT1 has also been explored. The most optimized glycolipid peptide R-C12-4 demonstrated significant cytotoxicity and antimetastasis in vitro and in vivo, which indicated that it may be a good lead for anticancer drug development.
Click to Show/Hide
|
||||
| Description |
For the subcutaneous transplantation model, R-C12 or R-C12-4 was injected intraperitoneally every other day at the dose of 15 mg/kg. As shown in Figure 7A,B, after 12 days, R-C12-4 and R-C12 have a significant inhibition effect on the BALB/c mice tumor volume compared to the control group (p < 0.01 and p < 0.05, respectively). For the metastasis model, R-C12 or R-C12-4 was applied by the tail intravenous injection every 2 days at the dose of 10 mg/kg. As shown in Figure 7D, significantly fewer metastatic nodules from the lungs of Kunming (KM) mice were observed in the R-C12-4 and R-C12 treatment groups compared to the control and this result was confirmed by the H&E staining of lung sections. Visible tumor nodules were counted on the surface of the lung demonstrating a significant difference between the control and R-C12 and R-C12-4 treatment groups (p < 0.05). It is noteworthy that both R-C12 and R-C12-4 did not affect the mouse body weight either in the subcutaneous transplantation model or in the metastasis model, indicating that these peptides did not diminish their overall health.
Click to Show/Hide
|
||||
| In Vivo Model | KM mice B16-F10 cells xenograft model. | ||||
| In Vitro Model | Mouse melanoma | B16-F10 cell | CVCL_0159 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
73.70%
|
|||
| Administration Time | 12 days | ||||
| Administration Dosage | 200 μL 15 mg/kg | ||||
| MOA of PDC |
In this study, we would investigate the co-modification of fatty acids and monosaccharides in anticancer peptides, as we expect to optimize the selectivity and cytotoxicity of the peptide at the same time. Based on our previous study, the lipopeptide R-C12 was selected as a template to perform glucose derivative modification at different positions as we supposed that the glucose derivative modification position in R-C12 might be a crucial factor in its selectivity and activity optimization. The glucose derivative was covalently coupled to different sites in R-C12 through its -amino group of a Lys residue, which was obtained by replacing the Arg residue. R-C12 contains seven Arg residues, accordingly, seven glycolipid peptides were designed and synthesized, which were named R-C12-1, R-C12-2, R-C12-3, R-C12-4, R-C12-5, R-C12-6, and R-C12-7. Various physiochemical properties including the hydrodynamic size, ζ-potential, secondary structure, and hydrophobicity were determined to explain the different cytotoxicity and selectivity of these glycolipid peptides. On the other hand, the strength of the binding between glycolipid peptides and the cancer cells mediated by GLUT1 has also been explored. The most optimized glycolipid peptide R-C12-4 demonstrated significant cytotoxicity and antimetastasis in vitro and in vivo, which indicated that it may be a good lead for anticancer drug development.
Click to Show/Hide
|
||||
| Description |
For the subcutaneous transplantation model, R-C12 or R-C12-4 was injected intraperitoneally every other day at the dose of 15 mg/kg. As shown in Figure 7A,B, after 12 days, R-C12-4 and R-C12 have a significant inhibition effect on the BALB/c mice tumor volume compared to the control group (p < 0.01 and p < 0.05, respectively). For the metastasis model, R-C12 or R-C12-4 was applied by the tail intravenous injection every 2 days at the dose of 10 mg/kg. As shown in Figure 7D, significantly fewer metastatic nodules from the lungs of Kunming (KM) mice were observed in the R-C12-4 and R-C12 treatment groups compared to the control and this result was confirmed by the H&E staining of lung sections. Visible tumor nodules were counted on the surface of the lung demonstrating a significant difference between the control and R-C12 and R-C12-4 treatment groups (p < 0.05). It is noteworthy that both R-C12 and R-C12-4 did not affect the mouse body weight either in the subcutaneous transplantation model or in the metastasis model, indicating that these peptides did not diminish their overall health.
Click to Show/Hide
|
||||
| In Vivo Model | KM mice B16-F10 cells xenograft model. | ||||
| In Vitro Model | Mouse melanoma | B16-F10 cell | CVCL_0159 | ||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
1.9 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, we would investigate the co-modification of fatty acids and monosaccharides in anticancer peptides, as we expect to optimize the selectivity and cytotoxicity of the peptide at the same time. Based on our previous study, the lipopeptide R-C12 was selected as a template to perform glucose derivative modification at different positions as we supposed that the glucose derivative modification position in R-C12 might be a crucial factor in its selectivity and activity optimization. The glucose derivative was covalently coupled to different sites in R-C12 through its -amino group of a Lys residue, which was obtained by replacing the Arg residue. R-C12 contains seven Arg residues, accordingly, seven glycolipid peptides were designed and synthesized, which were named R-C12-1, R-C12-2, R-C12-3, R-C12-4, R-C12-5, R-C12-6, and R-C12-7. Various physiochemical properties including the hydrodynamic size, ζ-potential, secondary structure, and hydrophobicity were determined to explain the different cytotoxicity and selectivity of these glycolipid peptides. On the other hand, the strength of the binding between glycolipid peptides and the cancer cells mediated by GLUT1 has also been explored. The most optimized glycolipid peptide R-C12-4 demonstrated significant cytotoxicity and antimetastasis in vitro and in vivo, which indicated that it may be a good lead for anticancer drug development.
Click to Show/Hide
|
||||
| Description |
Murine melanoma cells (B16-F10) were chosen as the object for evaluating the cytotoxicity of the seven glycolipid peptides and R-C12 by the CCK-8 assay. Based on our previous study, the HEK-293T cell line was also chosen as the noncancer cell line to evaluate the selectivity of glycolipid peptides. IC50 represents the concentration of glycolipid peptides and R-C12 that induce 50% cell death. As shown in Figure 1A, modification of the glucose derivative at different sites of R-C12 results in different cytotoxicity profiles. According to the IC50 values, for B16-F10 cells, the cytotoxic activity of peptides was arranged as follows R-C12-4 > R-C12-1 ≈ R-C12-2 ≈ R-C12-3 ≈ R-C12 > R-C12-5 > R-C12-6 > R-C12-7, in which R-C12-4 demonstrated the highest cytotoxicity on B16-F10 cells (IC50 = 1.9 μM)
Click to Show/Hide
|
||||
| In Vitro Model | Mouse melanoma | B16-F10 cell | CVCL_0159 | ||
R-L-HCPT [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Tumer volume |
700 mm3
|
|||
| Administration Time | 10 days | ||||
| MOA of PDC |
Our previous studies demonstrated that R-lycosin-I was a typical cationic anticancer peptide that contained a high relative abundance of positively charged arginine residues and possessed an amphiphilic character to allow its assembly into nanostructures in aqueous environments. Herein, we designed and synthesized a self-assembling anticancer conjugate in which R-lycosin-I covalently coupled with HCPT, a DNA topoisomerase I inhibitor through the glutamic anhydride linker. This conjugate (R-L-HCPT) could spontaneously associate into uniform 40-60 nm nanospheres in aqueous solution. The R-L-HCPT nanospheres exhibited excellent antitumor growth activity and antimetastatic efficacy compared with free HCPT or free R-lycosin-I in vitro and in vivo. Our study might provide new opportunities for the development of a self-assembling peptide-drug delivery system that could synergistically enhance anticancer outcomes.
Click to Show/Hide
|
||||
| Description |
Compared with the tumors in the control group with a final tumor volume of 3800 mm3, the tumors in the R-L-HCPT group grew much more slowly, and the mean tumor volume expanded from 200 to 700 mm3 within 10 days.
|
||||
| In Vivo Model | Murine melanoma B16-F10 xenograft model in BALB/c mice. | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Body weight |
31g
|
|||
| Administration Time | 10 days | ||||
| MOA of PDC |
Our previous studies demonstrated that R-lycosin-I was a typical cationic anticancer peptide that contained a high relative abundance of positively charged arginine residues and possessed an amphiphilic character to allow its assembly into nanostructures in aqueous environments. Herein, we designed and synthesized a self-assembling anticancer conjugate in which R-lycosin-I covalently coupled with HCPT, a DNA topoisomerase I inhibitor through the glutamic anhydride linker. This conjugate (R-L-HCPT) could spontaneously associate into uniform 40-60 nm nanospheres in aqueous solution. The R-L-HCPT nanospheres exhibited excellent antitumor growth activity and antimetastatic efficacy compared with free HCPT or free R-lycosin-I in vitro and in vivo. Our study might provide new opportunities for the development of a self-assembling peptide-drug delivery system that could synergistically enhance anticancer outcomes.
Click to Show/Hide
|
||||
| Description |
Notably, there were no significant body weight losses (Figure 6D), suggesting that R-L-HCPT did not induce systemic toxicity.
|
||||
| In Vivo Model | Murine melanoma B16-F10 xenograft model in BALB/c mice. | ||||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
1.1 μM
|
|||
| Administration Time | 48 h | ||||
| MOA of PDC |
Our previous studies demonstrated that R-lycosin-I was a typical cationic anticancer peptide that contained a high relative abundance of positively charged arginine residues and possessed an amphiphilic character to allow its assembly into nanostructures in aqueous environments. Herein, we designed and synthesized a self-assembling anticancer conjugate in which R-lycosin-I covalently coupled with HCPT, a DNA topoisomerase I inhibitor through the glutamic anhydride linker. This conjugate (R-L-HCPT) could spontaneously associate into uniform 40-60 nm nanospheres in aqueous solution. The R-L-HCPT nanospheres exhibited excellent antitumor growth activity and antimetastatic efficacy compared with free HCPT or free R-lycosin-I in vitro and in vivo. Our study might provide new opportunities for the development of a self-assembling peptide-drug delivery system that could synergistically enhance anticancer outcomes.
Click to Show/Hide
|
||||
| Description |
The cytotoxic activity of R-L-HCPT on MHCC-97H cells was time-dependent. With R-L-HCPT treatment for 6 and 12 h, the IC50 values were 13.5 μM and 11.7 μM, respectively. However, when the incubation time was extended to 24 and 48 h, the IC50 values were greatly reduced to 3.12 μM and 1.1 μM, respectively.
|
||||
| In Vitro Model | Hepatocellular carcinoma | MHCC97H cell | CVCL_4972 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
2.8 ± 0.4 μM
|
|||
| MOA of PDC |
Our previous studies demonstrated that R-lycosin-I was a typical cationic anticancer peptide that contained a high relative abundance of positively charged arginine residues and possessed an amphiphilic character to allow its assembly into nanostructures in aqueous environments. Herein, we designed and synthesized a self-assembling anticancer conjugate in which R-lycosin-I covalently coupled with HCPT, a DNA topoisomerase I inhibitor through the glutamic anhydride linker. This conjugate (R-L-HCPT) could spontaneously associate into uniform 40-60 nm nanospheres in aqueous solution. The R-L-HCPT nanospheres exhibited excellent antitumor growth activity and antimetastatic efficacy compared with free HCPT or free R-lycosin-I in vitro and in vivo. Our study might provide new opportunities for the development of a self-assembling peptide-drug delivery system that could synergistically enhance anticancer outcomes.
Click to Show/Hide
|
||||
| Description |
In addition, as shown in Table 1, the other cancer cell lines (HeLa, MDA-MB-231, B16-F10, HepG2, and A549) and noncancer cell lines (HEK293t, Beas-2B, and LO2) were evaluated in the cytotoxicity test. The IC50 values of R-L-HCPT were 2.9-6.6 μM for cancer cell lines and 2.8-5.8 μM for noncancer cell lines.
|
||||
| In Vitro Model | Amelanotic melanoma | LO #2 cell | CVCL_C7SD | ||
| Experiment 3 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
2.9 ± 0.5 μM
|
|||
| MOA of PDC |
Our previous studies demonstrated that R-lycosin-I was a typical cationic anticancer peptide that contained a high relative abundance of positively charged arginine residues and possessed an amphiphilic character to allow its assembly into nanostructures in aqueous environments. Herein, we designed and synthesized a self-assembling anticancer conjugate in which R-lycosin-I covalently coupled with HCPT, a DNA topoisomerase I inhibitor through the glutamic anhydride linker. This conjugate (R-L-HCPT) could spontaneously associate into uniform 40-60 nm nanospheres in aqueous solution. The R-L-HCPT nanospheres exhibited excellent antitumor growth activity and antimetastatic efficacy compared with free HCPT or free R-lycosin-I in vitro and in vivo. Our study might provide new opportunities for the development of a self-assembling peptide-drug delivery system that could synergistically enhance anticancer outcomes.
Click to Show/Hide
|
||||
| Description |
In addition, as shown in Table 1, the other cancer cell lines (HeLa, MDA-MB-231, B16-F10, HepG2, and A549) and noncancer cell lines (HEK293t, Beas-2B, and LO2) were evaluated in the cytotoxicity test. The IC50 values of R-L-HCPT were 2.9-6.6 μM for cancer cell lines and 2.8-5.8 μM for noncancer cell lines.
|
||||
| In Vitro Model | Hepatoblastoma | Hep-G2 cell | CVCL_0027 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
3.12 μM
|
|||
| MOA of PDC |
Our previous studies demonstrated that R-lycosin-I was a typical cationic anticancer peptide that contained a high relative abundance of positively charged arginine residues and possessed an amphiphilic character to allow its assembly into nanostructures in aqueous environments. Herein, we designed and synthesized a self-assembling anticancer conjugate in which R-lycosin-I covalently coupled with HCPT, a DNA topoisomerase I inhibitor through the glutamic anhydride linker. This conjugate (R-L-HCPT) could spontaneously associate into uniform 40-60 nm nanospheres in aqueous solution. The R-L-HCPT nanospheres exhibited excellent antitumor growth activity and antimetastatic efficacy compared with free HCPT or free R-lycosin-I in vitro and in vivo. Our study might provide new opportunities for the development of a self-assembling peptide-drug delivery system that could synergistically enhance anticancer outcomes.
Click to Show/Hide
|
||||
| Description |
The IC50 values for R-lycosin-I, R-L-HCPT conjugates, and HCPT were 15.27, 3.12, and 23.83 μM, respectively.
|
||||
| In Vitro Model | Hepatocellular carcinoma | MHCC97H cell | CVCL_4972 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
3.12 μM
|
|||
| Administration Time | 24 h | ||||
| MOA of PDC |
Our previous studies demonstrated that R-lycosin-I was a typical cationic anticancer peptide that contained a high relative abundance of positively charged arginine residues and possessed an amphiphilic character to allow its assembly into nanostructures in aqueous environments. Herein, we designed and synthesized a self-assembling anticancer conjugate in which R-lycosin-I covalently coupled with HCPT, a DNA topoisomerase I inhibitor through the glutamic anhydride linker. This conjugate (R-L-HCPT) could spontaneously associate into uniform 40-60 nm nanospheres in aqueous solution. The R-L-HCPT nanospheres exhibited excellent antitumor growth activity and antimetastatic efficacy compared with free HCPT or free R-lycosin-I in vitro and in vivo. Our study might provide new opportunities for the development of a self-assembling peptide-drug delivery system that could synergistically enhance anticancer outcomes.
Click to Show/Hide
|
||||
| Description |
The cytotoxic activity of R-L-HCPT on MHCC-97H cells was time-dependent. With R-L-HCPT treatment for 6 and 12 h, the IC50 values were 13.5 μM and 11.7 μM, respectively. However, when the incubation time was extended to 24 and 48 h, the IC50 values were greatly reduced to 3.12 μM and 1.1 μM, respectively.
|
||||
| In Vitro Model | Hepatocellular carcinoma | MHCC97H cell | CVCL_4972 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
3.9 ± 0.4 μM
|
|||
| MOA of PDC |
Our previous studies demonstrated that R-lycosin-I was a typical cationic anticancer peptide that contained a high relative abundance of positively charged arginine residues and possessed an amphiphilic character to allow its assembly into nanostructures in aqueous environments. Herein, we designed and synthesized a self-assembling anticancer conjugate in which R-lycosin-I covalently coupled with HCPT, a DNA topoisomerase I inhibitor through the glutamic anhydride linker. This conjugate (R-L-HCPT) could spontaneously associate into uniform 40-60 nm nanospheres in aqueous solution. The R-L-HCPT nanospheres exhibited excellent antitumor growth activity and antimetastatic efficacy compared with free HCPT or free R-lycosin-I in vitro and in vivo. Our study might provide new opportunities for the development of a self-assembling peptide-drug delivery system that could synergistically enhance anticancer outcomes.
Click to Show/Hide
|
||||
| Description |
In addition, as shown in Table 1, the other cancer cell lines (HeLa, MDA-MB-231, B16-F10, HepG2, and A549) and noncancer cell lines (HEK293t, Beas-2B, and LO2) were evaluated in the cytotoxicity test. The IC50 values of R-L-HCPT were 2.9-6.6 μM for cancer cell lines and 2.8-5.8 μM for noncancer cell lines.
|
||||
| In Vitro Model | Lung adenocarcinoma | A-549 cell | CVCL_0023 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
4.4 ± 0.5 μM
|
|||
| MOA of PDC |
Our previous studies demonstrated that R-lycosin-I was a typical cationic anticancer peptide that contained a high relative abundance of positively charged arginine residues and possessed an amphiphilic character to allow its assembly into nanostructures in aqueous environments. Herein, we designed and synthesized a self-assembling anticancer conjugate in which R-lycosin-I covalently coupled with HCPT, a DNA topoisomerase I inhibitor through the glutamic anhydride linker. This conjugate (R-L-HCPT) could spontaneously associate into uniform 40-60 nm nanospheres in aqueous solution. The R-L-HCPT nanospheres exhibited excellent antitumor growth activity and antimetastatic efficacy compared with free HCPT or free R-lycosin-I in vitro and in vivo. Our study might provide new opportunities for the development of a self-assembling peptide-drug delivery system that could synergistically enhance anticancer outcomes.
Click to Show/Hide
|
||||
| Description |
In addition, as shown in Table 1, the other cancer cell lines (HeLa, MDA-MB-231, B16-F10, HepG2, and A549) and noncancer cell lines (HEK293t, Beas-2B, and LO2) were evaluated in the cytotoxicity test. The IC50 values of R-L-HCPT were 2.9-6.6 μM for cancer cell lines and 2.8-5.8 μM for noncancer cell lines.
|
||||
| In Vitro Model | Normal | BEAS-2B cell | CVCL_0168 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
4.9 ± 0.3 μM
|
|||
| MOA of PDC |
Our previous studies demonstrated that R-lycosin-I was a typical cationic anticancer peptide that contained a high relative abundance of positively charged arginine residues and possessed an amphiphilic character to allow its assembly into nanostructures in aqueous environments. Herein, we designed and synthesized a self-assembling anticancer conjugate in which R-lycosin-I covalently coupled with HCPT, a DNA topoisomerase I inhibitor through the glutamic anhydride linker. This conjugate (R-L-HCPT) could spontaneously associate into uniform 40-60 nm nanospheres in aqueous solution. The R-L-HCPT nanospheres exhibited excellent antitumor growth activity and antimetastatic efficacy compared with free HCPT or free R-lycosin-I in vitro and in vivo. Our study might provide new opportunities for the development of a self-assembling peptide-drug delivery system that could synergistically enhance anticancer outcomes.
Click to Show/Hide
|
||||
| Description |
In addition, as shown in Table 1, the other cancer cell lines (HeLa, MDA-MB-231, B16-F10, HepG2, and A549) and noncancer cell lines (HEK293t, Beas-2B, and LO2) were evaluated in the cytotoxicity test. The IC50 values of R-L-HCPT were 2.9-6.6 μM for cancer cell lines and 2.8-5.8 μM for noncancer cell lines.
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
5.8 ± 1.1 μM
|
|||
| MOA of PDC |
Our previous studies demonstrated that R-lycosin-I was a typical cationic anticancer peptide that contained a high relative abundance of positively charged arginine residues and possessed an amphiphilic character to allow its assembly into nanostructures in aqueous environments. Herein, we designed and synthesized a self-assembling anticancer conjugate in which R-lycosin-I covalently coupled with HCPT, a DNA topoisomerase I inhibitor through the glutamic anhydride linker. This conjugate (R-L-HCPT) could spontaneously associate into uniform 40-60 nm nanospheres in aqueous solution. The R-L-HCPT nanospheres exhibited excellent antitumor growth activity and antimetastatic efficacy compared with free HCPT or free R-lycosin-I in vitro and in vivo. Our study might provide new opportunities for the development of a self-assembling peptide-drug delivery system that could synergistically enhance anticancer outcomes.
Click to Show/Hide
|
||||
| Description |
In addition, as shown in Table 1, the other cancer cell lines (HeLa, MDA-MB-231, B16-F10, HepG2, and A549) and noncancer cell lines (HEK293t, Beas-2B, and LO2) were evaluated in the cytotoxicity test. The IC50 values of R-L-HCPT were 2.9-6.6 μM for cancer cell lines and 2.8-5.8 μM for noncancer cell lines.
|
||||
| In Vitro Model | Normal | HEK-293T cell | CVCL_0063 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
6.1 ± 0.1 μM
|
|||
| MOA of PDC |
Our previous studies demonstrated that R-lycosin-I was a typical cationic anticancer peptide that contained a high relative abundance of positively charged arginine residues and possessed an amphiphilic character to allow its assembly into nanostructures in aqueous environments. Herein, we designed and synthesized a self-assembling anticancer conjugate in which R-lycosin-I covalently coupled with HCPT, a DNA topoisomerase I inhibitor through the glutamic anhydride linker. This conjugate (R-L-HCPT) could spontaneously associate into uniform 40-60 nm nanospheres in aqueous solution. The R-L-HCPT nanospheres exhibited excellent antitumor growth activity and antimetastatic efficacy compared with free HCPT or free R-lycosin-I in vitro and in vivo. Our study might provide new opportunities for the development of a self-assembling peptide-drug delivery system that could synergistically enhance anticancer outcomes.
Click to Show/Hide
|
||||
| Description |
In addition, as shown in Table 1, the other cancer cell lines (HeLa, MDA-MB-231, B16-F10, HepG2, and A549) and noncancer cell lines (HEK293t, Beas-2B, and LO2) were evaluated in the cytotoxicity test. The IC50 values of R-L-HCPT were 2.9-6.6 μM for cancer cell lines and 2.8-5.8 μM for noncancer cell lines.
|
||||
| In Vitro Model | Mouse melanoma | B16-F10 cell | CVCL_0159 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
6.6 ± 0.3 μM
|
|||
| MOA of PDC |
Our previous studies demonstrated that R-lycosin-I was a typical cationic anticancer peptide that contained a high relative abundance of positively charged arginine residues and possessed an amphiphilic character to allow its assembly into nanostructures in aqueous environments. Herein, we designed and synthesized a self-assembling anticancer conjugate in which R-lycosin-I covalently coupled with HCPT, a DNA topoisomerase I inhibitor through the glutamic anhydride linker. This conjugate (R-L-HCPT) could spontaneously associate into uniform 40-60 nm nanospheres in aqueous solution. The R-L-HCPT nanospheres exhibited excellent antitumor growth activity and antimetastatic efficacy compared with free HCPT or free R-lycosin-I in vitro and in vivo. Our study might provide new opportunities for the development of a self-assembling peptide-drug delivery system that could synergistically enhance anticancer outcomes.
Click to Show/Hide
|
||||
| Description |
In addition, as shown in Table 1, the other cancer cell lines (HeLa, MDA-MB-231, B16-F10, HepG2, and A549) and noncancer cell lines (HEK293t, Beas-2B, and LO2) were evaluated in the cytotoxicity test. The IC50 values of R-L-HCPT were 2.9-6.6 μM for cancer cell lines and 2.8-5.8 μM for noncancer cell lines.
|
||||
| In Vitro Model | Human papillomavirus-related cervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
11.7 μM
|
|||
| Administration Time | 12 h | ||||
| MOA of PDC |
Our previous studies demonstrated that R-lycosin-I was a typical cationic anticancer peptide that contained a high relative abundance of positively charged arginine residues and possessed an amphiphilic character to allow its assembly into nanostructures in aqueous environments. Herein, we designed and synthesized a self-assembling anticancer conjugate in which R-lycosin-I covalently coupled with HCPT, a DNA topoisomerase I inhibitor through the glutamic anhydride linker. This conjugate (R-L-HCPT) could spontaneously associate into uniform 40-60 nm nanospheres in aqueous solution. The R-L-HCPT nanospheres exhibited excellent antitumor growth activity and antimetastatic efficacy compared with free HCPT or free R-lycosin-I in vitro and in vivo. Our study might provide new opportunities for the development of a self-assembling peptide-drug delivery system that could synergistically enhance anticancer outcomes.
Click to Show/Hide
|
||||
| Description |
The cytotoxic activity of R-L-HCPT on MHCC-97H cells was time-dependent. With R-L-HCPT treatment for 6 and 12 h, the IC50 values were 13.5 μM and 11.7 μM, respectively. However, when the incubation time was extended to 24 and 48 h, the IC50 values were greatly reduced to 3.12 μM and 1.1 μM, respectively.
|
||||
| In Vitro Model | Hepatocellular carcinoma | MHCC97H cell | CVCL_4972 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
13.5 μM
|
|||
| Administration Time | 6 h | ||||
| MOA of PDC |
Our previous studies demonstrated that R-lycosin-I was a typical cationic anticancer peptide that contained a high relative abundance of positively charged arginine residues and possessed an amphiphilic character to allow its assembly into nanostructures in aqueous environments. Herein, we designed and synthesized a self-assembling anticancer conjugate in which R-lycosin-I covalently coupled with HCPT, a DNA topoisomerase I inhibitor through the glutamic anhydride linker. This conjugate (R-L-HCPT) could spontaneously associate into uniform 40-60 nm nanospheres in aqueous solution. The R-L-HCPT nanospheres exhibited excellent antitumor growth activity and antimetastatic efficacy compared with free HCPT or free R-lycosin-I in vitro and in vivo. Our study might provide new opportunities for the development of a self-assembling peptide-drug delivery system that could synergistically enhance anticancer outcomes.
Click to Show/Hide
|
||||
| Description |
The cytotoxic activity of R-L-HCPT on MHCC-97H cells was time-dependent. With R-L-HCPT treatment for 6 and 12 h, the IC50 values were 13.5 μM and 11.7 μM, respectively. However, when the incubation time was extended to 24 and 48 h, the IC50 values were greatly reduced to 3.12 μM and 1.1 μM, respectively.
|
||||
| In Vitro Model | Hepatocellular carcinoma | MHCC97H cell | CVCL_4972 | ||
E1-3 doxorubicin [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Medulloblastoma | ||||
| Efficacy Data | Blood-brain barrier permeability |
8
|
|||
| Administration Time | 30 min | ||||
| Description |
Notably, its permeability efficiency was significantly higher compared to free doxorubicin (5) (36.93 ± 0.7 uM and 28.93 ± 0.2 uM, respectively, p < 0.001) at 120 min post-treatment (Figure 8D).
|
||||
| In Vivo Model | Blood brain barrier model. | ||||
| In Vitro Model | Normal | HBEC-5i cell | CVCL_4D10 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Medulloblastoma | ||||
| Efficacy Data | Blood-brain barrier permeability |
20
|
|||
| Administration Time | 60 min | ||||
| Description |
Notably, its permeability efficiency was significantly higher compared to free doxorubicin (5) (36.93 ± 0.7 uM and 28.93 ± 0.2 uM, respectively, p < 0.001) at 120 min post-treatment (Figure 8D).
|
||||
| In Vivo Model | Blood brain barrier model. | ||||
| In Vitro Model | Normal | HBEC-5i cell | CVCL_4D10 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Medulloblastoma | ||||
| Efficacy Data | Blood-brain barrier permeability |
37
|
|||
| Administration Time | 120 min | ||||
| Description |
Notably, its permeability efficiency was significantly higher compared to free doxorubicin (5) (36.93 ± 0.7 uM and 28.93 ± 0.2 uM, respectively, p < 0.001) at 120 min post-treatment (Figure 8D).
|
||||
| In Vivo Model | Blood brain barrier model. | ||||
| In Vitro Model | Normal | HBEC-5i cell | CVCL_4D10 | ||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Medulloblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
25 ± 1.22 nM
|
|||
| Administration Time | 72 h | ||||
| Description |
This was confirmed with E1-7 doxorubicin conjugate (4) displaying a 5-fold reduction in cytotoxicity compared to E1-3 doxorubicin conjugate (3) (IC50 values of 130 ± 1.27 nM and 25 ± 1.22 nM, respectively) and 14-fold reduction in cytotoxicity compared to free doxorubicin (5) (IC50 values of 130 ± 1.27 nM and 8.8 ± 1.31 nM, respectively) (Figure 7).
Click to Show/Hide
|
||||
| In Vitro Model | Medulloblastoma | Medulloblastoma cell | Homo sapiens | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Medulloblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
842.0 ± 1.10 nM
|
|||
| Administration Time | 72 h | ||||
| Description |
E1-3 doxorubicin conjugate had a pronounced reduction in cytotoxicity (>72-fold reduction, IC50 value of 10754 ± 1.38 nM) compared to free doxorubicin (IC50 value of 148 ± 1.15 nM) in human fibroblasts. E1-7 doxorubicin was also able to reduce the cytotoxicity of doxorubicin on fibroblasts but not to the same degree as the E1-3 doxorubicin conjugate. E1-3 doxorubicin conjugate (3) also had reduced cytotoxicity compared to free doxorubicin (>7.4-fold reduction, IC50 values of 842 ± 1.10 nM and 113 ± 1.14 nM, respectively) in primary cultures of human astrocytes, a major cell type located in the brain and spinal cord.
Click to Show/Hide
|
||||
| In Vitro Model | Glioma | Brain astrocytes | Homo sapiens | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Medulloblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
10754 ± 1.38 nM
|
|||
| Administration Time | 72 h | ||||
| Description |
E1-3 doxorubicin conjugate had a pronounced reduction in cytotoxicity (>72-fold reduction, IC50 value of 10754 ± 1.38 nM) compared to free doxorubicin (IC50 value of 148 ± 1.15 nM) in human fibroblasts. E1-7 doxorubicin was also able to reduce the cytotoxicity of doxorubicin on fibroblasts but not to the same degree as the E1-3 doxorubicin conjugate. E1-3 doxorubicin conjugate (3) also had reduced cytotoxicity compared to free doxorubicin (>7.4-fold reduction, IC50 values of 842 ± 1.10 nM and 113 ± 1.14 nM, respectively) in primary cultures of human astrocytes, a major cell type located in the brain and spinal cord.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MRC-5 cell | CVCL_0440 | ||
Geo77 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
9 ± 0.4 µM
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Peptide-drug conjugates are delivery systems for selective delivery of cytotoxic agents to target cancer cells. In this work, the optimized synthesis of JH-VII-139-1 and its c(RGDyK) peptide conjugates is presented. The low nanomolar SRPK1 inhibitor, JH-VII-139-1, which is an analogue of Alectinib, was linked to the β3 targeting oligopeptide c(RGDyK) through amide, carbamate and urea linkers. The chemostability, cytotoxic and antiangiogenic properties of the synthesized hybrids were thoroughly studied. All conjugates retained mid nanomolar-level inhibitory activity against SRPK1 kinase and two out of four conjugates, geo75 and geo77 exhibited antiproliferative effects with low micromolar IC50 values against HeLa, K562, MDA-MB231 and MCF7 cancer cells. The activities were strongly related to the stability of the linkers and the release of JH-VII-139-1. In vivo zebrafish screening assays demonstrated the ability of the synthesized conjugates to inhibit the length or width of intersegmental vessels (ISVs). Flow cytometry experiments were used to test the cellular uptake of a fluorescein tagged hybrid in MCF7 and MDA-MB231 cells that revealed a receptor-mediated endocytosis process. In conclusion, most conjugates retained the inhibitory potency against SRPK1 as JH-VII-139-1 and demonstrated antiproliferative and antiangiogenic activities. Further animal model experiments are needed to uncover the full potential of such peptide conjugates in cancer therapy and angiogenesis-related diseases.
Click to Show/Hide
|
||||
| Description |
The cytotoxic activity of JH-VII-139-1 and JH-VII-139-1-c(RGDyK) hybrid compounds was evaluated at different concentrations (0.5-50 uM) over a panel of cell lines including HeLa cervical cancer, MCF7 mammary carcinoma, the triple-negative breast cancer MDA-MB-231 and K562 lymphoblast cells. Integrin receptors are highly overexpressed on the surface of many types of cancer. The metastatic breast cancer cell lines MDA-MB-435 and MCF-7, as well as HeLa cells, express high levels of V3 integrins. On the other hand, K562 cells express very low levels of V3 integrins. The cytotoxic and cytostatic activities of the JH-VII-139-1-c(RGDyK) hybrid compounds were estimated by three concentration-dependent parameters: GI50 (concentration that results in 50% growth inhibition), TGI (concentration that results in total growth inhibition or cytostatic effect), and IC50 (concentration that results in 50% growth cytotoxic effect). JH-VII-139-1 and the hybrid compounds, geo75 and geo77, showed a significant cytostatic (GI50 = 4-12 uM) and cytotoxic (IC50 = 3-18 uM) effect against HeLa, MCF-7, MDA-MB-231 and K562 cancer cells. The most active compound was geo77, showing IC50 and GI50 values of 3 uM and 4 uM, respectively in the most sensitive cell line MCF-7. Geo85 exhibited a slightly cytostatic effect (GI50 = 33 uM), with a minor cytotoxicity (IC50 = 45 uM) only against MCF-7 cells.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Half life period | 10-50 min | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Cervical carcinoma | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
11 ± 0.3 µM
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Peptide-drug conjugates are delivery systems for selective delivery of cytotoxic agents to target cancer cells. In this work, the optimized synthesis of JH-VII-139-1 and its c(RGDyK) peptide conjugates is presented. The low nanomolar SRPK1 inhibitor, JH-VII-139-1, which is an analogue of Alectinib, was linked to the β3 targeting oligopeptide c(RGDyK) through amide, carbamate and urea linkers. The chemostability, cytotoxic and antiangiogenic properties of the synthesized hybrids were thoroughly studied. All conjugates retained mid nanomolar-level inhibitory activity against SRPK1 kinase and two out of four conjugates, geo75 and geo77 exhibited antiproliferative effects with low micromolar IC50 values against HeLa, K562, MDA-MB231 and MCF7 cancer cells. The activities were strongly related to the stability of the linkers and the release of JH-VII-139-1. In vivo zebrafish screening assays demonstrated the ability of the synthesized conjugates to inhibit the length or width of intersegmental vessels (ISVs). Flow cytometry experiments were used to test the cellular uptake of a fluorescein tagged hybrid in MCF7 and MDA-MB231 cells that revealed a receptor-mediated endocytosis process. In conclusion, most conjugates retained the inhibitory potency against SRPK1 as JH-VII-139-1 and demonstrated antiproliferative and antiangiogenic activities. Further animal model experiments are needed to uncover the full potential of such peptide conjugates in cancer therapy and angiogenesis-related diseases.
Click to Show/Hide
|
||||
| Description |
The cytotoxic activity of JH-VII-139-1 and JH-VII-139-1-c(RGDyK) hybrid compounds was evaluated at different concentrations (0.5-50 uM) over a panel of cell lines including HeLa cervical cancer, MCF7 mammary carcinoma, the triple-negative breast cancer MDA-MB-231 and K562 lymphoblast cells. Integrin receptors are highly overexpressed on the surface of many types of cancer. The metastatic breast cancer cell lines MDA-MB-435 and MCF-7, as well as HeLa cells, express high levels of V3 integrins. On the other hand, K562 cells express very low levels of V3 integrins. The cytotoxic and cytostatic activities of the JH-VII-139-1-c(RGDyK) hybrid compounds were estimated by three concentration-dependent parameters: GI50 (concentration that results in 50% growth inhibition), TGI (concentration that results in total growth inhibition or cytostatic effect), and IC50 (concentration that results in 50% growth cytotoxic effect). JH-VII-139-1 and the hybrid compounds, geo75 and geo77, showed a significant cytostatic (GI50 = 4-12 uM) and cytotoxic (IC50 = 3-18 uM) effect against HeLa, MCF-7, MDA-MB-231 and K562 cancer cells. The most active compound was geo77, showing IC50 and GI50 values of 3 uM and 4 uM, respectively in the most sensitive cell line MCF-7. Geo85 exhibited a slightly cytostatic effect (GI50 = 33 uM), with a minor cytotoxicity (IC50 = 45 uM) only against MCF-7 cells.
Click to Show/Hide
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Half life period | 10-50 min | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
15 ± 0.7 µM
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Peptide-drug conjugates are delivery systems for selective delivery of cytotoxic agents to target cancer cells. In this work, the optimized synthesis of JH-VII-139-1 and its c(RGDyK) peptide conjugates is presented. The low nanomolar SRPK1 inhibitor, JH-VII-139-1, which is an analogue of Alectinib, was linked to the β3 targeting oligopeptide c(RGDyK) through amide, carbamate and urea linkers. The chemostability, cytotoxic and antiangiogenic properties of the synthesized hybrids were thoroughly studied. All conjugates retained mid nanomolar-level inhibitory activity against SRPK1 kinase and two out of four conjugates, geo75 and geo77 exhibited antiproliferative effects with low micromolar IC50 values against HeLa, K562, MDA-MB231 and MCF7 cancer cells. The activities were strongly related to the stability of the linkers and the release of JH-VII-139-1. In vivo zebrafish screening assays demonstrated the ability of the synthesized conjugates to inhibit the length or width of intersegmental vessels (ISVs). Flow cytometry experiments were used to test the cellular uptake of a fluorescein tagged hybrid in MCF7 and MDA-MB231 cells that revealed a receptor-mediated endocytosis process. In conclusion, most conjugates retained the inhibitory potency against SRPK1 as JH-VII-139-1 and demonstrated antiproliferative and antiangiogenic activities. Further animal model experiments are needed to uncover the full potential of such peptide conjugates in cancer therapy and angiogenesis-related diseases.
Click to Show/Hide
|
||||
| Description |
The cytotoxic activity of JH-VII-139-1 and JH-VII-139-1-c(RGDyK) hybrid compounds was evaluated at different concentrations (0.5-50 uM) over a panel of cell lines including HeLa cervical cancer, MCF7 mammary carcinoma, the triple-negative breast cancer MDA-MB-231 and K562 lymphoblast cells. Integrin receptors are highly overexpressed on the surface of many types of cancer. The metastatic breast cancer cell lines MDA-MB-435 and MCF-7, as well as HeLa cells, express high levels of V3 integrins. On the other hand, K562 cells express very low levels of V3 integrins. The cytotoxic and cytostatic activities of the JH-VII-139-1-c(RGDyK) hybrid compounds were estimated by three concentration-dependent parameters: GI50 (concentration that results in 50% growth inhibition), TGI (concentration that results in total growth inhibition or cytostatic effect), and IC50 (concentration that results in 50% growth cytotoxic effect). JH-VII-139-1 and the hybrid compounds, geo75 and geo77, showed a significant cytostatic (GI50 = 4-12 uM) and cytotoxic (IC50 = 3-18 uM) effect against HeLa, MCF-7, MDA-MB-231 and K562 cancer cells. The most active compound was geo77, showing IC50 and GI50 values of 3 uM and 4 uM, respectively in the most sensitive cell line MCF-7. Geo85 exhibited a slightly cytostatic effect (GI50 = 33 uM), with a minor cytotoxicity (IC50 = 45 uM) only against MCF-7 cells.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Half life period | 10-50 min | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Chronic myeloid leukemia | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
23 ± 0.5 µM
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Peptide-drug conjugates are delivery systems for selective delivery of cytotoxic agents to target cancer cells. In this work, the optimized synthesis of JH-VII-139-1 and its c(RGDyK) peptide conjugates is presented. The low nanomolar SRPK1 inhibitor, JH-VII-139-1, which is an analogue of Alectinib, was linked to the β3 targeting oligopeptide c(RGDyK) through amide, carbamate and urea linkers. The chemostability, cytotoxic and antiangiogenic properties of the synthesized hybrids were thoroughly studied. All conjugates retained mid nanomolar-level inhibitory activity against SRPK1 kinase and two out of four conjugates, geo75 and geo77 exhibited antiproliferative effects with low micromolar IC50 values against HeLa, K562, MDA-MB231 and MCF7 cancer cells. The activities were strongly related to the stability of the linkers and the release of JH-VII-139-1. In vivo zebrafish screening assays demonstrated the ability of the synthesized conjugates to inhibit the length or width of intersegmental vessels (ISVs). Flow cytometry experiments were used to test the cellular uptake of a fluorescein tagged hybrid in MCF7 and MDA-MB231 cells that revealed a receptor-mediated endocytosis process. In conclusion, most conjugates retained the inhibitory potency against SRPK1 as JH-VII-139-1 and demonstrated antiproliferative and antiangiogenic activities. Further animal model experiments are needed to uncover the full potential of such peptide conjugates in cancer therapy and angiogenesis-related diseases.
Click to Show/Hide
|
||||
| Description |
The cytotoxic activity of JH-VII-139-1 and JH-VII-139-1-c(RGDyK) hybrid compounds was evaluated at different concentrations (0.5-50 uM) over a panel of cell lines including HeLa cervical cancer, MCF7 mammary carcinoma, the triple-negative breast cancer MDA-MB-231 and K562 lymphoblast cells. Integrin receptors are highly overexpressed on the surface of many types of cancer. The metastatic breast cancer cell lines MDA-MB-435 and MCF-7, as well as HeLa cells, express high levels of V3 integrins. On the other hand, K562 cells express very low levels of V3 integrins. The cytotoxic and cytostatic activities of the JH-VII-139-1-c(RGDyK) hybrid compounds were estimated by three concentration-dependent parameters: GI50 (concentration that results in 50% growth inhibition), TGI (concentration that results in total growth inhibition or cytostatic effect), and IC50 (concentration that results in 50% growth cytotoxic effect). JH-VII-139-1 and the hybrid compounds, geo75 and geo77, showed a significant cytostatic (GI50 = 4-12 uM) and cytotoxic (IC50 = 3-18 uM) effect against HeLa, MCF-7, MDA-MB-231 and K562 cancer cells. The most active compound was geo77, showing IC50 and GI50 values of 3 uM and 4 uM, respectively in the most sensitive cell line MCF-7. Geo85 exhibited a slightly cytostatic effect (GI50 = 33 uM), with a minor cytotoxicity (IC50 = 45 uM) only against MCF-7 cells.
Click to Show/Hide
|
||||
| In Vitro Model | Chronic myeloid leukemia | K562 cell | CVCL_0004 | ||
| Half life period | 10-50 min | ||||
| Experiment 5 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
3 ± 0.4 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Peptide-drug conjugates are delivery systems for selective delivery of cytotoxic agents to target cancer cells. In this work, the optimized synthesis of JH-VII-139-1 and its c(RGDyK) peptide conjugates is presented. The low nanomolar SRPK1 inhibitor, JH-VII-139-1, which is an analogue of Alectinib, was linked to the β3targeting oligopeptide c(RGDyK) through amide, carbamate and urea linkers. The chemostability, cytotoxic and antiangiogenic properties of the synthesized hybrids were thoroughly studied. All conjugates retained mid nanomolar-level inhibitory activity against SRPK1 kinase and two out of four conjugates,geo75andgeo77exhibited antiproliferative effects with low micromolar IC50values against HeLa, K562, MDA-MB231 and MCF7 cancer cells. The activities were strongly related to the stability of the linkers and the release of JH-VII-139-1. In vivo zebrafish screening assays demonstrated the ability of the synthesized conjugates to inhibit the length or width of intersegmental vessels (ISVs). Flow cytometry experiments were used to test the cellular uptake of a fluorescein tagged hybrid in MCF7 and MDA-MB231 cells that revealed a receptor-mediated endocytosis process. In conclusion, most conjugates retained the inhibitory potency against SRPK1 as JH-VII-139-1 and demonstrated antiproliferative and antiangiogenic activities. Further animal model experiments are needed to uncover the full potential of such peptide conjugates in cancer therapy and angiogenesis-related diseases.
Click to Show/Hide
|
||||
| Description |
The cytotoxic activity of JH-VII-139-1 and JH-VII-139-1-c(RGDyK) hybrid compounds was evaluated at different concentrations (0.5-50 uM) over a panel of cell lines including HeLa cervical cancer, MCF7 mammary carcinoma, the triple-negative breast cancer MDA-MB-231 and K562 lymphoblast cells. Integrin receptors are highly overexpressed on the surface of many types of cancer. The metastatic breast cancer cell lines MDA-MB-435 and MCF-7, as well as HeLa cells, express high levels of V3 integrins. On the other hand, K562 cells express very low levels of V3 integrins. The cytotoxic and cytostatic activities of the JH-VII-139-1-c(RGDyK) hybrid compounds were estimated by three concentration-dependent parameters: GI50 (concentration that results in 50% growth inhibition), TGI (concentration that results in total growth inhibition or cytostatic effect), and IC50 (concentration that results in 50% growth cytotoxic effect). JH-VII-139-1 and the hybrid compounds, geo75 and geo77, showed a significant cytostatic (GI50 = 4-12 uM) and cytotoxic (IC50 = 3-18 uM) effect against HeLa, MCF-7, MDA-MB-231 and K562 cancer cells. The most active compound was geo77, showing IC50 and GI50 values of 3 uM and 4 uM, respectively in the most sensitive cell line MCF-7. Geo85 exhibited a slightly cytostatic effect (GI50 = 33 uM), with a minor cytotoxicity (IC50 = 45 uM) only against MCF-7 cells.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Half life period | 10-50 min | ||||
| Experiment 6 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Cervical carcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
9 ± 0.2 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Peptide-drug conjugates are delivery systems for selective delivery of cytotoxic agents to target cancer cells. In this work, the optimized synthesis of JH-VII-139-1 and its c(RGDyK) peptide conjugates is presented. The low nanomolar SRPK1 inhibitor, JH-VII-139-1, which is an analogue of Alectinib, was linked to the β3targeting oligopeptide c(RGDyK) through amide, carbamate and urea linkers. The chemostability, cytotoxic and antiangiogenic properties of the synthesized hybrids were thoroughly studied. All conjugates retained mid nanomolar-level inhibitory activity against SRPK1 kinase and two out of four conjugates,geo75andgeo77exhibited antiproliferative effects with low micromolar IC50values against HeLa, K562, MDA-MB231 and MCF7 cancer cells. The activities were strongly related to the stability of the linkers and the release of JH-VII-139-1. In vivo zebrafish screening assays demonstrated the ability of the synthesized conjugates to inhibit the length or width of intersegmental vessels (ISVs). Flow cytometry experiments were used to test the cellular uptake of a fluorescein tagged hybrid in MCF7 and MDA-MB231 cells that revealed a receptor-mediated endocytosis process. In conclusion, most conjugates retained the inhibitory potency against SRPK1 as JH-VII-139-1 and demonstrated antiproliferative and antiangiogenic activities. Further animal model experiments are needed to uncover the full potential of such peptide conjugates in cancer therapy and angiogenesis-related diseases.
Click to Show/Hide
|
||||
| Description |
The cytotoxic activity of JH-VII-139-1 and JH-VII-139-1-c(RGDyK) hybrid compounds was evaluated at different concentrations (0.5-50 uM) over a panel of cell lines including HeLa cervical cancer, MCF7 mammary carcinoma, the triple-negative breast cancer MDA-MB-231 and K562 lymphoblast cells. Integrin receptors are highly overexpressed on the surface of many types of cancer. The metastatic breast cancer cell lines MDA-MB-435 and MCF-7, as well as HeLa cells, express high levels of V3 integrins. On the other hand, K562 cells express very low levels of V3 integrins. The cytotoxic and cytostatic activities of the JH-VII-139-1-c(RGDyK) hybrid compounds were estimated by three concentration-dependent parameters: GI50 (concentration that results in 50% growth inhibition), TGI (concentration that results in total growth inhibition or cytostatic effect), and IC50 (concentration that results in 50% growth cytotoxic effect). JH-VII-139-1 and the hybrid compounds, geo75 and geo77, showed a significant cytostatic (GI50 = 4-12 uM) and cytotoxic (IC50 = 3-18 uM) effect against HeLa, MCF-7, MDA-MB-231 and K562 cancer cells. The most active compound was geo77, showing IC50 and GI50 values of 3 uM and 4 uM, respectively in the most sensitive cell line MCF-7. Geo85 exhibited a slightly cytostatic effect (GI50 = 33 uM), with a minor cytotoxicity (IC50 = 45 uM) only against MCF-7 cells.
Click to Show/Hide
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Half life period | 10-50 min | ||||
| Experiment 7 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
10 ± 0.3 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Peptide-drug conjugates are delivery systems for selective delivery of cytotoxic agents to target cancer cells. In this work, the optimized synthesis of JH-VII-139-1 and its c(RGDyK) peptide conjugates is presented. The low nanomolar SRPK1 inhibitor, JH-VII-139-1, which is an analogue of Alectinib, was linked to the β3targeting oligopeptide c(RGDyK) through amide, carbamate and urea linkers. The chemostability, cytotoxic and antiangiogenic properties of the synthesized hybrids were thoroughly studied. All conjugates retained mid nanomolar-level inhibitory activity against SRPK1 kinase and two out of four conjugates,geo75andgeo77exhibited antiproliferative effects with low micromolar IC50values against HeLa, K562, MDA-MB231 and MCF7 cancer cells. The activities were strongly related to the stability of the linkers and the release of JH-VII-139-1. In vivo zebrafish screening assays demonstrated the ability of the synthesized conjugates to inhibit the length or width of intersegmental vessels (ISVs). Flow cytometry experiments were used to test the cellular uptake of a fluorescein tagged hybrid in MCF7 and MDA-MB231 cells that revealed a receptor-mediated endocytosis process. In conclusion, most conjugates retained the inhibitory potency against SRPK1 as JH-VII-139-1 and demonstrated antiproliferative and antiangiogenic activities. Further animal model experiments are needed to uncover the full potential of such peptide conjugates in cancer therapy and angiogenesis-related diseases.
Click to Show/Hide
|
||||
| Description |
The cytotoxic activity of JH-VII-139-1 and JH-VII-139-1-c(RGDyK) hybrid compounds was evaluated at different concentrations (0.5-50 uM) over a panel of cell lines including HeLa cervical cancer, MCF7 mammary carcinoma, the triple-negative breast cancer MDA-MB-231 and K562 lymphoblast cells. Integrin receptors are highly overexpressed on the surface of many types of cancer. The metastatic breast cancer cell lines MDA-MB-435 and MCF-7, as well as HeLa cells, express high levels of V3 integrins. On the other hand, K562 cells express very low levels of V3 integrins. The cytotoxic and cytostatic activities of the JH-VII-139-1-c(RGDyK) hybrid compounds were estimated by three concentration-dependent parameters: GI50 (concentration that results in 50% growth inhibition), TGI (concentration that results in total growth inhibition or cytostatic effect), and IC50 (concentration that results in 50% growth cytotoxic effect). JH-VII-139-1 and the hybrid compounds, geo75 and geo77, showed a significant cytostatic (GI50 = 4-12 uM) and cytotoxic (IC50 = 3-18 uM) effect against HeLa, MCF-7, MDA-MB-231 and K562 cancer cells. The most active compound was geo77, showing IC50 and GI50 values of 3 uM and 4 uM, respectively in the most sensitive cell line MCF-7. Geo85 exhibited a slightly cytostatic effect (GI50 = 33 uM), with a minor cytotoxicity (IC50 = 45 uM) only against MCF-7 cells.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Half life period | 10-50 min | ||||
| Experiment 8 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Chronic myeloid leukemia | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
17 ± 0.5 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Peptide-drug conjugates are delivery systems for selective delivery of cytotoxic agents to target cancer cells. In this work, the optimized synthesis of JH-VII-139-1 and its c(RGDyK) peptide conjugates is presented. The low nanomolar SRPK1 inhibitor, JH-VII-139-1, which is an analogue of Alectinib, was linked to the β3targeting oligopeptide c(RGDyK) through amide, carbamate and urea linkers. The chemostability, cytotoxic and antiangiogenic properties of the synthesized hybrids were thoroughly studied. All conjugates retained mid nanomolar-level inhibitory activity against SRPK1 kinase and two out of four conjugates,geo75andgeo77exhibited antiproliferative effects with low micromolar IC50values against HeLa, K562, MDA-MB231 and MCF7 cancer cells. The activities were strongly related to the stability of the linkers and the release of JH-VII-139-1. In vivo zebrafish screening assays demonstrated the ability of the synthesized conjugates to inhibit the length or width of intersegmental vessels (ISVs). Flow cytometry experiments were used to test the cellular uptake of a fluorescein tagged hybrid in MCF7 and MDA-MB231 cells that revealed a receptor-mediated endocytosis process. In conclusion, most conjugates retained the inhibitory potency against SRPK1 as JH-VII-139-1 and demonstrated antiproliferative and antiangiogenic activities. Further animal model experiments are needed to uncover the full potential of such peptide conjugates in cancer therapy and angiogenesis-related diseases.
Click to Show/Hide
|
||||
| Description |
The cytotoxic activity of JH-VII-139-1 and JH-VII-139-1-c(RGDyK) hybrid compounds was evaluated at different concentrations (0.5-50 uM) over a panel of cell lines including HeLa cervical cancer, MCF7 mammary carcinoma, the triple-negative breast cancer MDA-MB-231 and K562 lymphoblast cells. Integrin receptors are highly overexpressed on the surface of many types of cancer. The metastatic breast cancer cell lines MDA-MB-435 and MCF-7, as well as HeLa cells, express high levels of V3 integrins. On the other hand, K562 cells express very low levels of V3 integrins. The cytotoxic and cytostatic activities of the JH-VII-139-1-c(RGDyK) hybrid compounds were estimated by three concentration-dependent parameters: GI50 (concentration that results in 50% growth inhibition), TGI (concentration that results in total growth inhibition or cytostatic effect), and IC50 (concentration that results in 50% growth cytotoxic effect). JH-VII-139-1 and the hybrid compounds, geo75 and geo77, showed a significant cytostatic (GI50 = 4-12 uM) and cytotoxic (IC50 = 3-18 uM) effect against HeLa, MCF-7, MDA-MB-231 and K562 cancer cells. The most active compound was geo77, showing IC50 and GI50 values of 3 uM and 4 uM, respectively in the most sensitive cell line MCF-7. Geo85 exhibited a slightly cytostatic effect (GI50 = 33 uM), with a minor cytotoxicity (IC50 = 45 uM) only against MCF-7 cells.
Click to Show/Hide
|
||||
| In Vitro Model | Chronic myeloid leukemia | K562 cell | CVCL_0004 | ||
| Half life period | 10-50 min | ||||
| Experiment 9 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal growth inhibition concentration (GI50) |
4 ± 0.4 µM
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Peptide-drug conjugates are delivery systems for selective delivery of cytotoxic agents to target cancer cells. In this work, the optimized synthesis of JH-VII-139-1 and its c(RGDyK) peptide conjugates is presented. The low nanomolar SRPK1 inhibitor, JH-VII-139-1, which is an analogue of Alectinib, was linked to the β3targeting oligopeptide c(RGDyK) through amide, carbamate and urea linkers. The chemostability, cytotoxic and antiangiogenic properties of the synthesized hybrids were thoroughly studied. All conjugates retained mid nanomolar-level inhibitory activity against SRPK1 kinase and two out of four conjugates,geo75andgeo77exhibited antiproliferative effects with low micromolar IC50values against HeLa, K562, MDA-MB231 and MCF7 cancer cells. The activities were strongly related to the stability of the linkers and the release of JH-VII-139-1. In vivo zebrafish screening assays demonstrated the ability of the synthesized conjugates to inhibit the length or width of intersegmental vessels (ISVs). Flow cytometry experiments were used to test the cellular uptake of a fluorescein tagged hybrid in MCF7 and MDA-MB231 cells that revealed a receptor-mediated endocytosis process. In conclusion, most conjugates retained the inhibitory potency against SRPK1 as JH-VII-139-1 and demonstrated antiproliferative and antiangiogenic activities. Further animal model experiments are needed to uncover the full potential of such peptide conjugates in cancer therapy and angiogenesis-related diseases.
Click to Show/Hide
|
||||
| Description |
The cytotoxic activity of JH-VII-139-1 and JH-VII-139-1-c(RGDyK) hybrid compounds was evaluated at different concentrations (0.5-50 uM) over a panel of cell lines including HeLa cervical cancer, MCF7 mammary carcinoma, the triple-negative breast cancer MDA-MB-231 and K562 lymphoblast cells. Integrin receptors are highly overexpressed on the surface of many types of cancer. The metastatic breast cancer cell lines MDA-MB-435 and MCF-7, as well as HeLa cells, express high levels of V3 integrins. On the other hand, K562 cells express very low levels of V3 integrins. The cytotoxic and cytostatic activities of the JH-VII-139-1-c(RGDyK) hybrid compounds were estimated by three concentration-dependent parameters: GI50 (concentration that results in 50% growth inhibition), TGI (concentration that results in total growth inhibition or cytostatic effect), and IC50 (concentration that results in 50% growth cytotoxic effect). JH-VII-139-1 and the hybrid compounds, geo75 and geo77, showed a significant cytostatic (GI50 = 4-12 uM) and cytotoxic (IC50 = 3-18 uM) effect against HeLa, MCF-7, MDA-MB-231 and K562 cancer cells. The most active compound was geo77, showing IC50 and GI50 values of 3 uM and 4 uM, respectively in the most sensitive cell line MCF-7. Geo85 exhibited a slightly cytostatic effect (GI50 = 33 uM), with a minor cytotoxicity (IC50 = 45 uM) only against MCF-7 cells.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Half life period | 10-50 min | ||||
| Experiment 10 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Cervical carcinoma | ||||
| Efficacy Data | Half Maximal growth inhibition concentration (GI50) |
5 ± 0.2 µM
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Peptide-drug conjugates are delivery systems for selective delivery of cytotoxic agents to target cancer cells. In this work, the optimized synthesis of JH-VII-139-1 and its c(RGDyK) peptide conjugates is presented. The low nanomolar SRPK1 inhibitor, JH-VII-139-1, which is an analogue of Alectinib, was linked to the β3targeting oligopeptide c(RGDyK) through amide, carbamate and urea linkers. The chemostability, cytotoxic and antiangiogenic properties of the synthesized hybrids were thoroughly studied. All conjugates retained mid nanomolar-level inhibitory activity against SRPK1 kinase and two out of four conjugates,geo75andgeo77exhibited antiproliferative effects with low micromolar IC50values against HeLa, K562, MDA-MB231 and MCF7 cancer cells. The activities were strongly related to the stability of the linkers and the release of JH-VII-139-1. In vivo zebrafish screening assays demonstrated the ability of the synthesized conjugates to inhibit the length or width of intersegmental vessels (ISVs). Flow cytometry experiments were used to test the cellular uptake of a fluorescein tagged hybrid in MCF7 and MDA-MB231 cells that revealed a receptor-mediated endocytosis process. In conclusion, most conjugates retained the inhibitory potency against SRPK1 as JH-VII-139-1 and demonstrated antiproliferative and antiangiogenic activities. Further animal model experiments are needed to uncover the full potential of such peptide conjugates in cancer therapy and angiogenesis-related diseases.
Click to Show/Hide
|
||||
| Description |
The cytotoxic activity of JH-VII-139-1 and JH-VII-139-1-c(RGDyK) hybrid compounds was evaluated at different concentrations (0.5-50 uM) over a panel of cell lines including HeLa cervical cancer, MCF7 mammary carcinoma, the triple-negative breast cancer MDA-MB-231 and K562 lymphoblast cells. Integrin receptors are highly overexpressed on the surface of many types of cancer. The metastatic breast cancer cell lines MDA-MB-435 and MCF-7, as well as HeLa cells, express high levels of V3 integrins. On the other hand, K562 cells express very low levels of V3 integrins. The cytotoxic and cytostatic activities of the JH-VII-139-1-c(RGDyK) hybrid compounds were estimated by three concentration-dependent parameters: GI50 (concentration that results in 50% growth inhibition), TGI (concentration that results in total growth inhibition or cytostatic effect), and IC50 (concentration that results in 50% growth cytotoxic effect). JH-VII-139-1 and the hybrid compounds, geo75 and geo77, showed a significant cytostatic (GI50 = 4-12 uM) and cytotoxic (IC50 = 3-18 uM) effect against HeLa, MCF-7, MDA-MB-231 and K562 cancer cells. The most active compound was geo77, showing IC50 and GI50 values of 3 uM and 4 uM, respectively in the most sensitive cell line MCF-7. Geo85 exhibited a slightly cytostatic effect (GI50 = 33 uM), with a minor cytotoxicity (IC50 = 45 uM) only against MCF-7 cells.
Click to Show/Hide
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Half life period | 10-50 min | ||||
| Experiment 11 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal growth inhibition concentration (GI50) |
8 ± 0.5 µM
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Peptide-drug conjugates are delivery systems for selective delivery of cytotoxic agents to target cancer cells. In this work, the optimized synthesis of JH-VII-139-1 and its c(RGDyK) peptide conjugates is presented. The low nanomolar SRPK1 inhibitor, JH-VII-139-1, which is an analogue of Alectinib, was linked to the β3targeting oligopeptide c(RGDyK) through amide, carbamate and urea linkers. The chemostability, cytotoxic and antiangiogenic properties of the synthesized hybrids were thoroughly studied. All conjugates retained mid nanomolar-level inhibitory activity against SRPK1 kinase and two out of four conjugates,geo75andgeo77exhibited antiproliferative effects with low micromolar IC50values against HeLa, K562, MDA-MB231 and MCF7 cancer cells. The activities were strongly related to the stability of the linkers and the release of JH-VII-139-1. In vivo zebrafish screening assays demonstrated the ability of the synthesized conjugates to inhibit the length or width of intersegmental vessels (ISVs). Flow cytometry experiments were used to test the cellular uptake of a fluorescein tagged hybrid in MCF7 and MDA-MB231 cells that revealed a receptor-mediated endocytosis process. In conclusion, most conjugates retained the inhibitory potency against SRPK1 as JH-VII-139-1 and demonstrated antiproliferative and antiangiogenic activities. Further animal model experiments are needed to uncover the full potential of such peptide conjugates in cancer therapy and angiogenesis-related diseases.
Click to Show/Hide
|
||||
| Description |
The cytotoxic activity of JH-VII-139-1 and JH-VII-139-1-c(RGDyK) hybrid compounds was evaluated at different concentrations (0.5-50 uM) over a panel of cell lines including HeLa cervical cancer, MCF7 mammary carcinoma, the triple-negative breast cancer MDA-MB-231 and K562 lymphoblast cells. Integrin receptors are highly overexpressed on the surface of many types of cancer. The metastatic breast cancer cell lines MDA-MB-435 and MCF-7, as well as HeLa cells, express high levels of V3 integrins. On the other hand, K562 cells express very low levels of V3 integrins. The cytotoxic and cytostatic activities of the JH-VII-139-1-c(RGDyK) hybrid compounds were estimated by three concentration-dependent parameters: GI50 (concentration that results in 50% growth inhibition), TGI (concentration that results in total growth inhibition or cytostatic effect), and IC50 (concentration that results in 50% growth cytotoxic effect). JH-VII-139-1 and the hybrid compounds, geo75 and geo77, showed a significant cytostatic (GI50 = 4-12 uM) and cytotoxic (IC50 = 3-18 uM) effect against HeLa, MCF-7, MDA-MB-231 and K562 cancer cells. The most active compound was geo77, showing IC50 and GI50 values of 3 uM and 4 uM, respectively in the most sensitive cell line MCF-7. Geo85 exhibited a slightly cytostatic effect (GI50 = 33 uM), with a minor cytotoxicity (IC50 = 45 uM) only against MCF-7 cells.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Half life period | 10-50 min | ||||
| Experiment 12 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Chronic myeloid leukemia | ||||
| Efficacy Data | Half Maximal growth inhibition concentration (GI50) |
12 ± 0.9 µM
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Peptide-drug conjugates are delivery systems for selective delivery of cytotoxic agents to target cancer cells. In this work, the optimized synthesis of JH-VII-139-1 and its c(RGDyK) peptide conjugates is presented. The low nanomolar SRPK1 inhibitor, JH-VII-139-1, which is an analogue of Alectinib, was linked to the β3targeting oligopeptide c(RGDyK) through amide, carbamate and urea linkers. The chemostability, cytotoxic and antiangiogenic properties of the synthesized hybrids were thoroughly studied. All conjugates retained mid nanomolar-level inhibitory activity against SRPK1 kinase and two out of four conjugates,geo75andgeo77exhibited antiproliferative effects with low micromolar IC50values against HeLa, K562, MDA-MB231 and MCF7 cancer cells. The activities were strongly related to the stability of the linkers and the release of JH-VII-139-1. In vivo zebrafish screening assays demonstrated the ability of the synthesized conjugates to inhibit the length or width of intersegmental vessels (ISVs). Flow cytometry experiments were used to test the cellular uptake of a fluorescein tagged hybrid in MCF7 and MDA-MB231 cells that revealed a receptor-mediated endocytosis process. In conclusion, most conjugates retained the inhibitory potency against SRPK1 as JH-VII-139-1 and demonstrated antiproliferative and antiangiogenic activities. Further animal model experiments are needed to uncover the full potential of such peptide conjugates in cancer therapy and angiogenesis-related diseases.
Click to Show/Hide
|
||||
| Description |
The cytotoxic activity of JH-VII-139-1 and JH-VII-139-1-c(RGDyK) hybrid compounds was evaluated at different concentrations (0.5-50 uM) over a panel of cell lines including HeLa cervical cancer, MCF7 mammary carcinoma, the triple-negative breast cancer MDA-MB-231 and K562 lymphoblast cells. Integrin receptors are highly overexpressed on the surface of many types of cancer. The metastatic breast cancer cell lines MDA-MB-435 and MCF-7, as well as HeLa cells, express high levels of V3 integrins. On the other hand, K562 cells express very low levels of V3 integrins. The cytotoxic and cytostatic activities of the JH-VII-139-1-c(RGDyK) hybrid compounds were estimated by three concentration-dependent parameters: GI50 (concentration that results in 50% growth inhibition), TGI (concentration that results in total growth inhibition or cytostatic effect), and IC50 (concentration that results in 50% growth cytotoxic effect). JH-VII-139-1 and the hybrid compounds, geo75 and geo77, showed a significant cytostatic (GI50 = 4-12 uM) and cytotoxic (IC50 = 3-18 uM) effect against HeLa, MCF-7, MDA-MB-231 and K562 cancer cells. The most active compound was geo77, showing IC50 and GI50 values of 3 uM and 4 uM, respectively in the most sensitive cell line MCF-7. Geo85 exhibited a slightly cytostatic effect (GI50 = 33 uM), with a minor cytotoxicity (IC50 = 45 uM) only against MCF-7 cells.
Click to Show/Hide
|
||||
| In Vitro Model | Chronic myeloid leukemia | K562 cell | CVCL_0004 | ||
| Half life period | 10-50 min | ||||
FLCpOH-TP10-NH2 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
8 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
15 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Hepatocellular carcinoma | MHCC97H cell | CVCL_4972 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
31 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | MIA PaCa-2 (KRAS G12C) cell | CVCL_0428 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
45 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MT4/HIV-1 cell | CVCL_RW54 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
45 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 (ACE+) cell | CVCL_0062 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
45 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Breast cancer | MCF-7/6 cell | CVCL_W972 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
45 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7/ADR cell | CVCL_0031 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
45 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive ductal carcinoma | MCF7/PTX cell | CVCL_C5RS | ||
| Experiment 9 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
45 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 (KRAS G13D) cell | CVCL_0062 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
45 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Uterine sarcoma | MES-SA/Dx5 cell | CVCL_2598 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
62 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Mouse colon adenocarcinoma | MC-38 cell | CVCL_B288 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
62 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
62 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Acute monocytic leukemia | MONO-MAC-6 cell | CVCL_1426 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
62 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 15 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Logarithmic survival |
1.02 CFU/ml
|
|||
| Administration Time | 6 h | ||||
| Administration Dosage | 4 MFC | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | T acute lymphoblastic leukemia | Jurkat cell | CVCL_0065 | ||
| Experiment 16 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Logarithmic survival |
1.08 CFU/ml
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 4 MFC | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | T acute lymphoblastic leukemia | Jurkat cell | CVCL_0065 | ||
| Experiment 17 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Logarithmic survival |
2.95 CFU/ml
|
|||
| Administration Time | 6 h | ||||
| Administration Dosage | 2 x MFC | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | T acute lymphoblastic leukemia | Jurkat cell | CVCL_0065 | ||
| Experiment 18 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Logarithmic survival |
3.76 CFU/ml
|
|||
| Administration Time | 4 h | ||||
| Administration Dosage | 2 x MFC | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | T acute lymphoblastic leukemia | Jurkat cell | CVCL_0065 | ||
| Experiment 19 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Logarithmic survival |
3.76 CFU/ml
|
|||
| Administration Time | 4 h | ||||
| Administration Dosage | 4 MFC | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | T acute lymphoblastic leukemia | Jurkat cell | CVCL_0065 | ||
| Experiment 20 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Logarithmic survival |
4.09 CFU/ml
|
|||
| Administration Time | 2 h | ||||
| Administration Dosage | 1 x MFC | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | T acute lymphoblastic leukemia | Jurkat cell | CVCL_0065 | ||
| Experiment 21 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Logarithmic survival |
4.12 CFU/ml
|
|||
| Administration Time | 6 h | ||||
| Administration Dosage | 1 x MFC | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | T acute lymphoblastic leukemia | Jurkat cell | CVCL_0065 | ||
| Experiment 22 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Logarithmic survival |
4.16 CFU/ml
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 1 x MFC | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | T acute lymphoblastic leukemia | Jurkat cell | CVCL_0065 | ||
| Experiment 23 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Logarithmic survival |
4.18 CFU/ml
|
|||
| Administration Time | 4 h | ||||
| Administration Dosage | 1 x MFC | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | T acute lymphoblastic leukemia | Jurkat cell | CVCL_0065 | ||
| Experiment 24 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Logarithmic survival |
4.66 CFU/ml
|
|||
| Administration Time | 2 h | ||||
| Administration Dosage | 2 x MFC | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | T acute lymphoblastic leukemia | Jurkat cell | CVCL_0065 | ||
| Experiment 25 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Logarithmic survival |
4.66 CFU/ml
|
|||
| Administration Time | 2 h | ||||
| Administration Dosage | 4 MFC | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | T acute lymphoblastic leukemia | Jurkat cell | CVCL_0065 | ||
| Experiment 26 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Logarithmic survival |
5.01 CFU/ml
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 2 x MFC | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | T acute lymphoblastic leukemia | Jurkat cell | CVCL_0065 | ||
| Experiment 27 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
15 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 28 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
31 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Hepatocellular carcinoma | MHCC97H cell | CVCL_4972 | ||
| Experiment 29 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
62 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | MIA PaCa-2 (KRAS G12C) cell | CVCL_0428 | ||
| Experiment 30 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
62 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MT4/HIV-1 cell | CVCL_RW54 | ||
| Experiment 31 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
62 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 (ACE+) cell | CVCL_0062 | ||
| Experiment 32 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
62 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Breast cancer | MCF-7/6 cell | CVCL_W972 | ||
| Experiment 33 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
62 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7/ADR cell | CVCL_0031 | ||
| Experiment 34 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
62 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive ductal carcinoma | MCF7/PTX cell | CVCL_C5RS | ||
| Experiment 35 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
62 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 (KRAS G13D) cell | CVCL_0062 | ||
| Experiment 36 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
62 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Uterine sarcoma | MES-SA/Dx5 cell | CVCL_2598 | ||
| Experiment 37 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
125 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Mouse colon adenocarcinoma | MC-38 cell | CVCL_B288 | ||
| Experiment 38 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
125 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
| Experiment 39 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
125 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Acute monocytic leukemia | MONO-MAC-6 cell | CVCL_1426 | ||
| Experiment 40 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
125 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 41 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
13.07 ± 0.35 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 42 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
15.16 ± 1.26 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Human umbilical vein endothelial cells | Homo sapiens | ||
| Experiment 43 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | 90% minimum inhibitory concentration (MIC90) |
31 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 44 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | 90% minimum inhibitory concentration (MIC90) |
62 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Hepatocellular carcinoma | MHCC97H cell | CVCL_4972 | ||
| Experiment 45 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | 90% minimum inhibitory concentration (MIC90) |
62 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | MIA PaCa-2 (KRAS G12C) cell | CVCL_0428 | ||
| Experiment 46 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | 90% minimum inhibitory concentration (MIC90) |
62 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MT4/HIV-1 cell | CVCL_RW54 | ||
| Experiment 47 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | 90% minimum inhibitory concentration (MIC90) |
62 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 48 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | 90% minimum inhibitory concentration (MIC90) |
125 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Acute monocytic leukemia | MONO-MAC-6 cell | CVCL_1426 | ||
| Experiment 49 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | 90% minimum inhibitory concentration (MIC90) |
250 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
| Experiment 50 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | 90% minimum inhibitory concentration (MIC90) |
250 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 (ACE+) cell | CVCL_0062 | ||
| Experiment 51 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | 90% minimum inhibitory concentration (MIC90) |
250 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Breast cancer | MCF-7/6 cell | CVCL_W972 | ||
| Experiment 52 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | 90% minimum inhibitory concentration (MIC90) |
250 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7/ADR cell | CVCL_0031 | ||
| Experiment 53 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | 90% minimum inhibitory concentration (MIC90) |
250 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive ductal carcinoma | MCF7/PTX cell | CVCL_C5RS | ||
| Experiment 54 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | 90% minimum inhibitory concentration (MIC90) |
250 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Uterine sarcoma | MES-SA/Dx5 cell | CVCL_2598 | ||
| Experiment 55 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | 90% maximal inhibitory concentration (IC50) |
29.78 ± 0.29 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 56 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | 90% maximal inhibitory concentration (IC50) |
34.03 ± 1.90 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Human umbilical vein endothelial cells | Homo sapiens | ||
FLCpOH-TP10-7-NH2 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
15 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
31 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MT4/HIV-1 cell | CVCL_RW54 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
62 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
62 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Hepatocellular carcinoma | MHCC97H cell | CVCL_4972 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
62 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | MIA PaCa-2 (KRAS G12C) cell | CVCL_0428 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
62 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Acute monocytic leukemia | MONO-MAC-6 cell | CVCL_1426 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
62 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
125 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 (ACE+) cell | CVCL_0062 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
125 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Breast cancer | MCF-7/6 cell | CVCL_W972 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
125 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7/ADR cell | CVCL_0031 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
125 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive ductal carcinoma | MCF7/PTX cell | CVCL_C5RS | ||
| Experiment 12 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
125 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 (KRAS G13D) cell | CVCL_0062 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
125 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Uterine sarcoma | MES-SA/Dx5 cell | CVCL_2598 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Logarithmic survival |
1.75 CFU/ml
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 4 MFC | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | T acute lymphoblastic leukemia | Jurkat cell | CVCL_0065 | ||
| Experiment 15 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Logarithmic survival |
2.86 CFU/ml
|
|||
| Administration Time | 6 h | ||||
| Administration Dosage | 2 x MFC | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | T acute lymphoblastic leukemia | Jurkat cell | CVCL_0065 | ||
| Experiment 16 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Logarithmic survival |
2.86 CFU/ml
|
|||
| Administration Time | 6 h | ||||
| Administration Dosage | 4 MFC | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | T acute lymphoblastic leukemia | Jurkat cell | CVCL_0065 | ||
| Experiment 17 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Logarithmic survival |
3.35 CFU/ml
|
|||
| Administration Time | 4 h | ||||
| Administration Dosage | 4 MFC | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | T acute lymphoblastic leukemia | Jurkat cell | CVCL_0065 | ||
| Experiment 18 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Logarithmic survival |
3.85 CFU/ml
|
|||
| Administration Time | 4 h | ||||
| Administration Dosage | 2 x MFC | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | T acute lymphoblastic leukemia | Jurkat cell | CVCL_0065 | ||
| Experiment 19 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Logarithmic survival |
4.32 CFU/ml
|
|||
| Administration Time | 2 h | ||||
| Administration Dosage | 4 MFC | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | T acute lymphoblastic leukemia | Jurkat cell | CVCL_0065 | ||
| Experiment 20 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Logarithmic survival |
4.66 CFU/ml
|
|||
| Administration Time | 6 h | ||||
| Administration Dosage | 1 x MFC | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | T acute lymphoblastic leukemia | Jurkat cell | CVCL_0065 | ||
| Experiment 21 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Logarithmic survival |
4.69 CFU/ml
|
|||
| Administration Time | 2 h | ||||
| Administration Dosage | 2 x MFC | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | T acute lymphoblastic leukemia | Jurkat cell | CVCL_0065 | ||
| Experiment 22 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Logarithmic survival |
4.7 CFU/ml
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 1 x MFC | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | T acute lymphoblastic leukemia | Jurkat cell | CVCL_0065 | ||
| Experiment 23 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Logarithmic survival |
4.73 CFU/ml
|
|||
| Administration Time | 4 h | ||||
| Administration Dosage | 1 x MFC | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | T acute lymphoblastic leukemia | Jurkat cell | CVCL_0065 | ||
| Experiment 24 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Logarithmic survival |
4.75 CFU/ml
|
|||
| Administration Time | 2 h | ||||
| Administration Dosage | 1 x MFC | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | T acute lymphoblastic leukemia | Jurkat cell | CVCL_0065 | ||
| Experiment 25 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Logarithmic survival |
5.36 CFU/ml
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 2 x MFC | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | T acute lymphoblastic leukemia | Jurkat cell | CVCL_0065 | ||
| Experiment 26 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
8 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 27 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
15 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Hepatocellular carcinoma | MHCC97H cell | CVCL_4972 | ||
| Experiment 28 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
15 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 (ACE+) cell | CVCL_0062 | ||
| Experiment 29 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
23 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MT4/HIV-1 cell | CVCL_RW54 | ||
| Experiment 30 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
23 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Breast cancer | MCF-7/6 cell | CVCL_W972 | ||
| Experiment 31 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
23 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7/ADR cell | CVCL_0031 | ||
| Experiment 32 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
23 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive ductal carcinoma | MCF7/PTX cell | CVCL_C5RS | ||
| Experiment 33 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
23 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 (KRAS G13D) cell | CVCL_0062 | ||
| Experiment 34 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
23 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Uterine sarcoma | MES-SA/Dx5 cell | CVCL_2598 | ||
| Experiment 35 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
31 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
| Experiment 36 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
31 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | MIA PaCa-2 (KRAS G12C) cell | CVCL_0428 | ||
| Experiment 37 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
31 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 38 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
45 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Acute monocytic leukemia | MONO-MAC-6 cell | CVCL_1426 | ||
| Experiment 39 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
190 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Mouse colon adenocarcinoma | MC-38 cell | CVCL_B288 | ||
| Experiment 40 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
12.00 ± 0.93 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 41 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
14.45 ± 1.13 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Human umbilical vein endothelial cells | Homo sapiens | ||
| Experiment 42 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | 90% minimum inhibitory concentration (MIC90) |
15 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 43 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | 90% minimum inhibitory concentration (MIC90) |
31 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Hepatocellular carcinoma | MHCC97H cell | CVCL_4972 | ||
| Experiment 44 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | 90% minimum inhibitory concentration (MIC90) |
31 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MT4/HIV-1 cell | CVCL_RW54 | ||
| Experiment 45 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | 90% minimum inhibitory concentration (MIC90) |
31 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 (ACE+) cell | CVCL_0062 | ||
| Experiment 46 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | 90% minimum inhibitory concentration (MIC90) |
31 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Breast cancer | MCF-7/6 cell | CVCL_W972 | ||
| Experiment 47 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | 90% minimum inhibitory concentration (MIC90) |
31 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7/ADR cell | CVCL_0031 | ||
| Experiment 48 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | 90% minimum inhibitory concentration (MIC90) |
31 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive ductal carcinoma | MCF7/PTX cell | CVCL_C5RS | ||
| Experiment 49 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | 90% minimum inhibitory concentration (MIC90) |
31 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 (KRAS G13D) cell | CVCL_0062 | ||
| Experiment 50 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | 90% minimum inhibitory concentration (MIC90) |
31 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Uterine sarcoma | MES-SA/Dx5 cell | CVCL_2598 | ||
| Experiment 51 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | 90% minimum inhibitory concentration (MIC90) |
62 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
| Experiment 52 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | 90% minimum inhibitory concentration (MIC90) |
62 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | MIA PaCa-2 (KRAS G12C) cell | CVCL_0428 | ||
| Experiment 53 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | 90% minimum inhibitory concentration (MIC90) |
62 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Acute monocytic leukemia | MONO-MAC-6 cell | CVCL_1426 | ||
| Experiment 54 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | 90% minimum inhibitory concentration (MIC90) |
62 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 55 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | 90% minimum inhibitory concentration (MIC90) |
250 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Mouse colon adenocarcinoma | MC-38 cell | CVCL_B288 | ||
| Experiment 56 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | 90% maximal inhibitory concentration (IC50) |
28.81 ± 0.87 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 57 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | 90% maximal inhibitory concentration (IC50) |
34.30 ± 1.00 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Human umbilical vein endothelial cells | Homo sapiens | ||
FLCpOH-LFcinB[Nle1,11]-NH2 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
23 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
23 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Breast cancer | MCF-7/6 cell | CVCL_W972 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
31 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
45 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Hepatocellular carcinoma | MHCC97H cell | CVCL_4972 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
45 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | MIA PaCa-2 (KRAS G12C) cell | CVCL_0428 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
45 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive ductal carcinoma | MCF7/PTX cell | CVCL_C5RS | ||
| Experiment 7 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
45 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 (KRAS G13D) cell | CVCL_0062 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
45 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Uterine sarcoma | MES-SA/Dx5 cell | CVCL_2598 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
90 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7/ADR cell | CVCL_0031 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
125 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MT4/HIV-1 cell | CVCL_RW54 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
250 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Mouse colon adenocarcinoma | MC-38 cell | CVCL_B288 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
250 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Acute monocytic leukemia | MONO-MAC-6 cell | CVCL_1426 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration | > 250 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
250 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 (ACE+) cell | CVCL_0062 | ||
| Experiment 15 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
31 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 16 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
31 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Breast cancer | MCF-7/6 cell | CVCL_W972 | ||
| Experiment 17 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
62 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
| Experiment 18 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
62 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Hepatocellular carcinoma | MHCC97H cell | CVCL_4972 | ||
| Experiment 19 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
62 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | MIA PaCa-2 (KRAS G12C) cell | CVCL_0428 | ||
| Experiment 20 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
62 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive ductal carcinoma | MCF7/PTX cell | CVCL_C5RS | ||
| Experiment 21 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
62 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 (KRAS G13D) cell | CVCL_0062 | ||
| Experiment 22 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
90 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Uterine sarcoma | MES-SA/Dx5 cell | CVCL_2598 | ||
| Experiment 23 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
125 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7/ADR cell | CVCL_0031 | ||
| Experiment 24 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) | > 250 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Mouse colon adenocarcinoma | MC-38 cell | CVCL_B288 | ||
| Experiment 25 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) | > 250 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Acute monocytic leukemia | MONO-MAC-6 cell | CVCL_1426 | ||
| Experiment 26 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
250 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MT4/HIV-1 cell | CVCL_RW54 | ||
| Experiment 27 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) | > 250 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 28 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) | > 250 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 (ACE+) cell | CVCL_0062 | ||
| Experiment 29 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
75.58 ± 1.73 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Human umbilical vein endothelial cells | Homo sapiens | ||
| Experiment 30 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | 90% minimum inhibitory concentration (MIC90) |
250 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 31 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | 90% maximal inhibitory concentration (IC50) | > 100 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 32 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | 90% maximal inhibitory concentration (IC50) | > 100 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 33 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | 90% maximal inhibitory concentration (IC50) | > 100 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Human umbilical vein endothelial cells | Homo sapiens | ||
FLCpOH-LFcinB(2-11)-NH2 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
31 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
62 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
125 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Hepatocellular carcinoma | MHCC97H cell | CVCL_4972 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
125 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | MIA PaCa-2 (KRAS G12C) cell | CVCL_0428 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
190 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Acute monocytic leukemia | MONO-MAC-6 cell | CVCL_1426 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
190 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Breast cancer | MCF-7/6 cell | CVCL_W972 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
190 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7/ADR cell | CVCL_0031 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
190 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive ductal carcinoma | MCF7/PTX cell | CVCL_C5RS | ||
| Experiment 9 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
190 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 (KRAS G13D) cell | CVCL_0062 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
190 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Uterine sarcoma | MES-SA/Dx5 cell | CVCL_2598 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration | > 250 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Mouse colon adenocarcinoma | MC-38 cell | CVCL_B288 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration | > 250 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MT4/HIV-1 cell | CVCL_RW54 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration | > 250 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
250 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 (ACE+) cell | CVCL_0062 | ||
| Experiment 15 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
62 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
| Experiment 16 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
125 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 17 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) | > 250 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Mouse colon adenocarcinoma | MC-38 cell | CVCL_B288 | ||
| Experiment 18 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
250 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Hepatocellular carcinoma | MHCC97H cell | CVCL_4972 | ||
| Experiment 19 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
250 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | MIA PaCa-2 (KRAS G12C) cell | CVCL_0428 | ||
| Experiment 20 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
250 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Acute monocytic leukemia | MONO-MAC-6 cell | CVCL_1426 | ||
| Experiment 21 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) | > 250 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MT4/HIV-1 cell | CVCL_RW54 | ||
| Experiment 22 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) | > 250 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 23 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) | > 250 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 (ACE+) cell | CVCL_0062 | ||
| Experiment 24 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
250 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Breast cancer | MCF-7/6 cell | CVCL_W972 | ||
| Experiment 25 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
250 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7/ADR cell | CVCL_0031 | ||
| Experiment 26 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
250 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive ductal carcinoma | MCF7/PTX cell | CVCL_C5RS | ||
| Experiment 27 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
250 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 (KRAS G13D) cell | CVCL_0062 | ||
| Experiment 28 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
250 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Uterine sarcoma | MES-SA/Dx5 cell | CVCL_2598 | ||
| Experiment 29 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 100 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 30 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 100 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Human umbilical vein endothelial cells | Homo sapiens | ||
| Experiment 31 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | 90% minimum inhibitory concentration (MIC90) |
125 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
| Experiment 32 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | 90% minimum inhibitory concentration (MIC90) |
250 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 33 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | 90% maximal inhibitory concentration (IC50) | > 100 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 34 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | 90% maximal inhibitory concentration (IC50) | > 100 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Human umbilical vein endothelial cells | Homo sapiens | ||
FLCpOH-HLopt2-NH2 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
62 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
125 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Hepatocellular carcinoma | MHCC97H cell | CVCL_4972 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration | > 250 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Mouse colon adenocarcinoma | MC-38 cell | CVCL_B288 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration | > 250 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration | > 250 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | MIA PaCa-2 (KRAS G12C) cell | CVCL_0428 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration | > 250 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Acute monocytic leukemia | MONO-MAC-6 cell | CVCL_1426 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration | > 250 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MT4/HIV-1 cell | CVCL_RW54 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration | > 250 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
250 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 (ACE+) cell | CVCL_0062 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration | > 250 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Breast cancer | MCF-7/6 cell | CVCL_W972 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
250 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7/ADR cell | CVCL_0031 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration | > 250 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive ductal carcinoma | MCF7/PTX cell | CVCL_C5RS | ||
| Experiment 13 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration |
250 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 (KRAS G13D) cell | CVCL_0062 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Minimum fungicidal concentration | > 250 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Uterine sarcoma | MES-SA/Dx5 cell | CVCL_2598 | ||
| Experiment 15 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
125 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 16 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) | > 250 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Mouse colon adenocarcinoma | MC-38 cell | CVCL_B288 | ||
| Experiment 17 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) | > 250 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
| Experiment 18 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) |
250 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Hepatocellular carcinoma | MHCC97H cell | CVCL_4972 | ||
| Experiment 19 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) | > 250 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | MIA PaCa-2 (KRAS G12C) cell | CVCL_0428 | ||
| Experiment 20 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) | > 250 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Acute monocytic leukemia | MONO-MAC-6 cell | CVCL_1426 | ||
| Experiment 21 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) | > 250 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MT4/HIV-1 cell | CVCL_RW54 | ||
| Experiment 22 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) | > 250 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 23 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) | > 250 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 (ACE+) cell | CVCL_0062 | ||
| Experiment 24 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) | > 250 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Breast cancer | MCF-7/6 cell | CVCL_W972 | ||
| Experiment 25 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) | > 250 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7/ADR cell | CVCL_0031 | ||
| Experiment 26 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) | > 250 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive ductal carcinoma | MCF7/PTX cell | CVCL_C5RS | ||
| Experiment 27 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) | > 250 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 (KRAS G13D) cell | CVCL_0062 | ||
| Experiment 28 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half minimum inhibitory concentration (MIC90) | > 250 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | Antifungal activity of the tested compounds against yeast cells was determined in 96-well plates, with minor modifications (CLSI, 2008). Suspensions of the microorganisms were prepared by taking one loop of pure culture into sterile water and adjusting optical density to 0.1 at 660 nm wavelength and further 50-fold dilution in RPMI 1640 medium, resulting in cell concentration of approximately 2 104 CFU/m. Then, 100 μL of cells were added to the wells of a 96-well microtiter plate that contained 16-500 μM tested compounds and 16-500 μM of FLC or 0.2-62 μM AmB. The plates were incubated for 24 h at 37 °C. Each test was performed in triplicate. The final concentration of DMSO was ensured to be around 1% in all experiments. The MIC90 was defined as the lowest concentration of drug showing no visible growth; MIC50 was defined as the concentration that yielded at least 50% growth inhibition when compared with the growth control well. For determination of minimum fungicidal concentrations (MFC), small aliquots of suspensions (around 10 μL) from each well were transferred using the pipette to YPD agar plates without inhibitors and incubated for 24 h at 37 °C. The MFC was defined as the lowest concentration of drug at which no growth of the colonies was observed. | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The inactivity of FLCpOH confirmed that the drug derivative, used during the synthesis of our target compounds, did not exert any antifungal activity. All compounds displayed poor activity against the C. glabrata strain tested. Another identified advantage of FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 is their higher fungicidal potential compared to fluconazole. The MFC values against C. albicans reference strains were the same or only twice higher compared to MIC90. The high fungicidal activity of both conjugates was also confirmed with the kill-time assay. However, at least a 4 x MFC concentration was necessary to obtain an evident killing effect after 24 h of treatment.
Click to Show/Hide
|
||||
| In Vitro Model | Uterine sarcoma | MES-SA/Dx5 cell | CVCL_2598 | ||
| Experiment 29 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 100 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 30 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 100 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Human umbilical vein endothelial cells | Homo sapiens | ||
| Experiment 31 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | 90% maximal inhibitory concentration (IC50) | > 100 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 32 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | <i>Candida spp</i> infection | ||||
| Efficacy Data | 90% maximal inhibitory concentration (IC50) | > 100 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Herein we report the chemical synthesis and biological activity of conjugates of fluconazole with (i) cell-penetrating peptides (CPP), namely TP10-NH2 and TP10-7-NH2, or (ii) antimicrobial peptides (AMP), such as LFcinB(2-11)-NH2, LFcinB[Nle1,11]-NH2 and HLopt2-NH2. Both constituents of produced conjugates (FLC and peptides) display different modes of antifungal activity and affect different molecular targets within fungal cells. CPPs serving as carrier peptides and are considered the fundamental part of an extensively developed concept of a drug delivery system; TP-10 is one of the most promising peptides in this family. These peptides effectively penetrate cell membrane. In our recent paper. we showed that TP10-NH2 can be successfully applied to design conjugates with antimicrobial activity. Modified fragments of bovine lactoferrin (LFcin) and human lactoferrin (HLopt2) are members of the AMP family, peptides that constitute an important segment of the bodys natural immunity against microorganisms.
Click to Show/Hide
|
||||
| Description |
The adverse drug effects associated with the use of antimicrobials can be a major concern especially with antifungal agents due to the eukaryotic nature of both the organism being targeted and the host. Therefore, it was important to test the toxicity of novel fluconazole-based conjugates. To examine and compare the cytotoxicity of FLC and its derivatives, the cytotoxicities of the compounds were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in two mammalian cell lines: human foreskin fibroblast cell line (Hs27) and human umbilical venous endothelial primary cells (HUVEC). The results showed that the FLC appeared to be less cytotoxic than its conjugates (Table 3). Among conjugates, FLCpOH-TP10-NH2 and FLCpOH-TP10-7-NH2 appeared to be more cytotoxic. However, the IC90 value for the human cells after 72 h treatment was comparable to the MIC50 value after 24 h treatment for most strains of C. albicans (Table S2).
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Human umbilical vein endothelial cells | Homo sapiens | ||
cCPP-CBT [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [6] | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
106 ± 8.3 nM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTS assay | ||||
| MOA of PDC |
The present study explores two different targeting peptides (TPs), RGDC (TP1), and CTVRTSAD (TP2), toward avβ1 integrin and EDB-Fn, respectively. TP1 or TP2 is covalently conjugated via disulfide bond to [C(WR)2K(WR)2]-(CBT) (6) to provide CBT conjugates (11, 16) containing an active targeting ability with increased aqueous solubility without decreasing cellular uptake. Targeting peptides may enable cCPP-CBT to target biomarker overexpressing tumor cells, and cCPP can assist in improving the physicochemical properties of CBT and delivery into the cancer cells. The conjugates in vitro stability was examined in various relevant conditions to mimic the cancerous versus normal physiological environment. Conjugates were evaluated in human plasma at two different glutathione concentrations (GSH, 1 mM and 10 mM) and two different pHs (6.5 and 7.4). The reported GSH level in noncancerous conditions is below 1 mM and approximately 10 mM at the cancer site. The in vitro antiproliferative assay using a biomarker overexpressing cell lines was compared to those without or with moderately expressing cancer cell lines. The study results demonstrate that targeting enhances the efficacy of CBT on cancer cell lines overexpressing extracellular biomarkers.
Click to Show/Hide
|
||||
| Description |
The conjugates therapeutic efficacy with targeting moieties was evaluated using prostate cancer cell lines with and without EDB-Fn overexpression (C4-2 and DU-145). Breast cancer cell lines with and without integrin v1 overexpression (MCF-7 and MDA-MB-231) were also used to evaluate antiproliferative activity. Noncancerous embryonic kidney epithelial cell line (HEK-293) was used as a control since this cell line did not express both biomarkers. All the compounds with and without targeting moieties showed comparable or lower antiproliferative activity than the CBT alone after 72 h incubation. The antiproliferative activity of compound 11 on MCF-7 (IC50 = 160.4 nM) was 1.6 times greater than on MDA-MB-231 (IC50 = 251.6 nM), showing a slight improvement in antiproliferative activity. A similar observation was found with compound 16 that showed 1.45 times higher antiproliferative activity on MCF-7 (IC50 = 162.4 nM) as compared to MDA-MB-231 cells (IC50 = 235.0 nM). Both MCF-7 and MDA-MB-231 are breast cancer cell lines. MDA-MB-231 is triple-negative breast cancer cells, and it showed higher IC50 values for CBT than MCF-7 cells. Later, the antiproliferative activity was compared between MCF-7 and the control cells (HEK-293); it was observed that compound 11 showed 3.33 times higher antiproliferative activity on MCF-7 cells (IC50 for 11 = 160.4 nM) compared to HEK-293 (IC50 for 11 = 534.5 nM), and compound 16 showed 4.6 times higher antiproliferative activity on MCF-7 (IC50 for 16 = 162.4 nM) compared to HEK-293 (IC50 for 16 = 483.3 nM). Thereafter, we wanted to assess the effectiveness of the EDB-Fn targeting moiety by comparing the antiproliferative activities on prostate cancer cell lines with and without EDB-Fn overexpression. The data showed a similar trend to that observed in breast cancer cells. For instance, compound 11 showed 3.4 times higher antiproliferative activity on C4-2 (IC50 for 11 = 181.4 nM) compared to DU-145 (IC50 for 11 = 611.2 nM). Similarly, compound 16 showed 3.7 times higher antiproliferative activity on C4-2 (IC50 for 16 = 202.2 nM) compared to DU-145 (IC50 for 16 = 751.1 nM). Compound 16 (possessing EDB-Fn targeting moiety) showed 1.1 times less antiproliferative activity compared to compound 6 (cCPP-CBT, without targeting moiety) on C4-2 cell lines, but compound 16 showed a much higher difference (3.6 times) in antiproliferative activity when compared with compound 6 on EDB-Fn moderately expressing DU-145 cell line. This data is further corroborated in EDB-Fn nonexpressing HEK-293. For instance, compound 16 showed 4.6 times less antiproliferative activity when compared with compound 6 on HEK-293 cell line. It can be inferred from the data (Table 2) that the EDB-Fn targeting conjugate was significantly effective on EDB-Fn overexpressing cells, while the conjugate has minimal effect on nonexpressing or moderately expressing cells. In conclusion, it is plausible to state that the presence of conjugating moieties on compound 11 (integrin targeting) and compound 16 (EDB-Fn targeting) significantly affects their ability to adhere and deliver the chemotherapeutic agent selectively to biomarker overexpressing cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | HEK293 cell | CVCL_0045 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Prostate carcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
177.4 ± 10.2 nM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTS assay | ||||
| MOA of PDC |
The present study explores two different targeting peptides (TPs), RGDC (TP1), and CTVRTSAD (TP2), toward avβ1 integrin and EDB-Fn, respectively. TP1 or TP2 is covalently conjugated via disulfide bond to [C(WR)2K(WR)2]-(CBT) (6) to provide CBT conjugates (11, 16) containing an active targeting ability with increased aqueous solubility without decreasing cellular uptake. Targeting peptides may enable cCPP-CBT to target biomarker overexpressing tumor cells, and cCPP can assist in improving the physicochemical properties of CBT and delivery into the cancer cells. The conjugates in vitro stability was examined in various relevant conditions to mimic the cancerous versus normal physiological environment. Conjugates were evaluated in human plasma at two different glutathione concentrations (GSH, 1 mM and 10 mM) and two different pHs (6.5 and 7.4). The reported GSH level in noncancerous conditions is below 1 mM and approximately 10 mM at the cancer site. The in vitro antiproliferative assay using a biomarker overexpressing cell lines was compared to those without or with moderately expressing cancer cell lines. The study results demonstrate that targeting enhances the efficacy of CBT on cancer cell lines overexpressing extracellular biomarkers.
Click to Show/Hide
|
||||
| Description |
The conjugates therapeutic efficacy with targeting moieties was evaluated using prostate cancer cell lines with and without EDB-Fn overexpression (C4-2 and DU-145). Breast cancer cell lines with and without integrin v1 overexpression (MCF-7 and MDA-MB-231) were also used to evaluate antiproliferative activity. Noncancerous embryonic kidney epithelial cell line (HEK-293) was used as a control since this cell line did not express both biomarkers. All the compounds with and without targeting moieties showed comparable or lower antiproliferative activity than the CBT alone after 72 h incubation. The antiproliferative activity of compound 11 on MCF-7 (IC50 = 160.4 nM) was 1.6 times greater than on MDA-MB-231 (IC50 = 251.6 nM), showing a slight improvement in antiproliferative activity. A similar observation was found with compound 16 that showed 1.45 times higher antiproliferative activity on MCF-7 (IC50 = 162.4 nM) as compared to MDA-MB-231 cells (IC50 = 235.0 nM). Both MCF-7 and MDA-MB-231 are breast cancer cell lines. MDA-MB-231 is triple-negative breast cancer cells, and it showed higher IC50 values for CBT than MCF-7 cells. Later, the antiproliferative activity was compared between MCF-7 and the control cells (HEK-293); it was observed that compound 11 showed 3.33 times higher antiproliferative activity on MCF-7 cells (IC50 for 11 = 160.4 nM) compared to HEK-293 (IC50 for 11 = 534.5 nM), and compound 16 showed 4.6 times higher antiproliferative activity on MCF-7 (IC50 for 16 = 162.4 nM) compared to HEK-293 (IC50 for 16 = 483.3 nM). Thereafter, we wanted to assess the effectiveness of the EDB-Fn targeting moiety by comparing the antiproliferative activities on prostate cancer cell lines with and without EDB-Fn overexpression. The data showed a similar trend to that observed in breast cancer cells. For instance, compound 11 showed 3.4 times higher antiproliferative activity on C4-2 (IC50 for 11 = 181.4 nM) compared to DU-145 (IC50 for 11 = 611.2 nM). Similarly, compound 16 showed 3.7 times higher antiproliferative activity on C4-2 (IC50 for 16 = 202.2 nM) compared to DU-145 (IC50 for 16 = 751.1 nM). Compound 16 (possessing EDB-Fn targeting moiety) showed 1.1 times less antiproliferative activity compared to compound 6 (cCPP-CBT, without targeting moiety) on C4-2 cell lines, but compound 16 showed a much higher difference (3.6 times) in antiproliferative activity when compared with compound 6 on EDB-Fn moderately expressing DU-145 cell line. This data is further corroborated in EDB-Fn nonexpressing HEK-293. For instance, compound 16 showed 4.6 times less antiproliferative activity when compared with compound 6 on HEK-293 cell line. It can be inferred from the data (Table 2) that the EDB-Fn targeting conjugate was significantly effective on EDB-Fn overexpressing cells, while the conjugate has minimal effect on nonexpressing or moderately expressing cells. In conclusion, it is plausible to state that the presence of conjugating moieties on compound 11 (integrin targeting) and compound 16 (EDB-Fn targeting) significantly affects their ability to adhere and deliver the chemotherapeutic agent selectively to biomarker overexpressing cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | LNCaP C4-2 cell | CVCL_4782 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Invasive breast carcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
205.3 ± 18.7 nM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTS assay | ||||
| MOA of PDC |
The present study explores two different targeting peptides (TPs), RGDC (TP1), and CTVRTSAD (TP2), toward avβ1 integrin and EDB-Fn, respectively. TP1 or TP2 is covalently conjugated via disulfide bond to [C(WR)2K(WR)2]-(CBT) (6) to provide CBT conjugates (11, 16) containing an active targeting ability with increased aqueous solubility without decreasing cellular uptake. Targeting peptides may enable cCPP-CBT to target biomarker overexpressing tumor cells, and cCPP can assist in improving the physicochemical properties of CBT and delivery into the cancer cells. The conjugates in vitro stability was examined in various relevant conditions to mimic the cancerous versus normal physiological environment. Conjugates were evaluated in human plasma at two different glutathione concentrations (GSH, 1 mM and 10 mM) and two different pHs (6.5 and 7.4). The reported GSH level in noncancerous conditions is below 1 mM and approximately 10 mM at the cancer site. The in vitro antiproliferative assay using a biomarker overexpressing cell lines was compared to those without or with moderately expressing cancer cell lines. The study results demonstrate that targeting enhances the efficacy of CBT on cancer cell lines overexpressing extracellular biomarkers.
Click to Show/Hide
|
||||
| Description |
The conjugates therapeutic efficacy with targeting moieties was evaluated using prostate cancer cell lines with and without EDB-Fn overexpression (C4-2 and DU-145). Breast cancer cell lines with and without integrin v1 overexpression (MCF-7 and MDA-MB-231) were also used to evaluate antiproliferative activity. Noncancerous embryonic kidney epithelial cell line (HEK-293) was used as a control since this cell line did not express both biomarkers. All the compounds with and without targeting moieties showed comparable or lower antiproliferative activity than the CBT alone after 72 h incubation. The antiproliferative activity of compound 11 on MCF-7 (IC50 = 160.4 nM) was 1.6 times greater than on MDA-MB-231 (IC50 = 251.6 nM), showing a slight improvement in antiproliferative activity. A similar observation was found with compound 16 that showed 1.45 times higher antiproliferative activity on MCF-7 (IC50 = 162.4 nM) as compared to MDA-MB-231 cells (IC50 = 235.0 nM). Both MCF-7 and MDA-MB-231 are breast cancer cell lines. MDA-MB-231 is triple-negative breast cancer cells, and it showed higher IC50 values for CBT than MCF-7 cells. Later, the antiproliferative activity was compared between MCF-7 and the control cells (HEK-293); it was observed that compound 11 showed 3.33 times higher antiproliferative activity on MCF-7 cells (IC50 for 11 = 160.4 nM) compared to HEK-293 (IC50 for 11 = 534.5 nM), and compound 16 showed 4.6 times higher antiproliferative activity on MCF-7 (IC50 for 16 = 162.4 nM) compared to HEK-293 (IC50 for 16 = 483.3 nM). Thereafter, we wanted to assess the effectiveness of the EDB-Fn targeting moiety by comparing the antiproliferative activities on prostate cancer cell lines with and without EDB-Fn overexpression. The data showed a similar trend to that observed in breast cancer cells. For instance, compound 11 showed 3.4 times higher antiproliferative activity on C4-2 (IC50 for 11 = 181.4 nM) compared to DU-145 (IC50 for 11 = 611.2 nM). Similarly, compound 16 showed 3.7 times higher antiproliferative activity on C4-2 (IC50 for 16 = 202.2 nM) compared to DU-145 (IC50 for 16 = 751.1 nM). Compound 16 (possessing EDB-Fn targeting moiety) showed 1.1 times less antiproliferative activity compared to compound 6 (cCPP-CBT, without targeting moiety) on C4-2 cell lines, but compound 16 showed a much higher difference (3.6 times) in antiproliferative activity when compared with compound 6 on EDB-Fn moderately expressing DU-145 cell line. This data is further corroborated in EDB-Fn nonexpressing HEK-293. For instance, compound 16 showed 4.6 times less antiproliferative activity when compared with compound 6 on HEK-293 cell line. It can be inferred from the data (Table 2) that the EDB-Fn targeting conjugate was significantly effective on EDB-Fn overexpressing cells, while the conjugate has minimal effect on nonexpressing or moderately expressing cells. In conclusion, it is plausible to state that the presence of conjugating moieties on compound 11 (integrin targeting) and compound 16 (EDB-Fn targeting) significantly affects their ability to adhere and deliver the chemotherapeutic agent selectively to biomarker overexpressing cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Prostate carcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
208.7 ± 8.7 nM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTS assay | ||||
| MOA of PDC |
The present study explores two different targeting peptides (TPs), RGDC (TP1), and CTVRTSAD (TP2), toward avβ1 integrin and EDB-Fn, respectively. TP1 or TP2 is covalently conjugated via disulfide bond to [C(WR)2K(WR)2]-(CBT) (6) to provide CBT conjugates (11, 16) containing an active targeting ability with increased aqueous solubility without decreasing cellular uptake. Targeting peptides may enable cCPP-CBT to target biomarker overexpressing tumor cells, and cCPP can assist in improving the physicochemical properties of CBT and delivery into the cancer cells. The conjugates in vitro stability was examined in various relevant conditions to mimic the cancerous versus normal physiological environment. Conjugates were evaluated in human plasma at two different glutathione concentrations (GSH, 1 mM and 10 mM) and two different pHs (6.5 and 7.4). The reported GSH level in noncancerous conditions is below 1 mM and approximately 10 mM at the cancer site. The in vitro antiproliferative assay using a biomarker overexpressing cell lines was compared to those without or with moderately expressing cancer cell lines. The study results demonstrate that targeting enhances the efficacy of CBT on cancer cell lines overexpressing extracellular biomarkers.
Click to Show/Hide
|
||||
| Description |
The conjugates therapeutic efficacy with targeting moieties was evaluated using prostate cancer cell lines with and without EDB-Fn overexpression (C4-2 and DU-145). Breast cancer cell lines with and without integrin v1 overexpression (MCF-7 and MDA-MB-231) were also used to evaluate antiproliferative activity. Noncancerous embryonic kidney epithelial cell line (HEK-293) was used as a control since this cell line did not express both biomarkers. All the compounds with and without targeting moieties showed comparable or lower antiproliferative activity than the CBT alone after 72 h incubation. The antiproliferative activity of compound 11 on MCF-7 (IC50 = 160.4 nM) was 1.6 times greater than on MDA-MB-231 (IC50 = 251.6 nM), showing a slight improvement in antiproliferative activity. A similar observation was found with compound 16 that showed 1.45 times higher antiproliferative activity on MCF-7 (IC50 = 162.4 nM) as compared to MDA-MB-231 cells (IC50 = 235.0 nM). Both MCF-7 and MDA-MB-231 are breast cancer cell lines. MDA-MB-231 is triple-negative breast cancer cells, and it showed higher IC50 values for CBT than MCF-7 cells. Later, the antiproliferative activity was compared between MCF-7 and the control cells (HEK-293); it was observed that compound 11 showed 3.33 times higher antiproliferative activity on MCF-7 cells (IC50 for 11 = 160.4 nM) compared to HEK-293 (IC50 for 11 = 534.5 nM), and compound 16 showed 4.6 times higher antiproliferative activity on MCF-7 (IC50 for 16 = 162.4 nM) compared to HEK-293 (IC50 for 16 = 483.3 nM). Thereafter, we wanted to assess the effectiveness of the EDB-Fn targeting moiety by comparing the antiproliferative activities on prostate cancer cell lines with and without EDB-Fn overexpression. The data showed a similar trend to that observed in breast cancer cells. For instance, compound 11 showed 3.4 times higher antiproliferative activity on C4-2 (IC50 for 11 = 181.4 nM) compared to DU-145 (IC50 for 11 = 611.2 nM). Similarly, compound 16 showed 3.7 times higher antiproliferative activity on C4-2 (IC50 for 16 = 202.2 nM) compared to DU-145 (IC50 for 16 = 751.1 nM). Compound 16 (possessing EDB-Fn targeting moiety) showed 1.1 times less antiproliferative activity compared to compound 6 (cCPP-CBT, without targeting moiety) on C4-2 cell lines, but compound 16 showed a much higher difference (3.6 times) in antiproliferative activity when compared with compound 6 on EDB-Fn moderately expressing DU-145 cell line. This data is further corroborated in EDB-Fn nonexpressing HEK-293. For instance, compound 16 showed 4.6 times less antiproliferative activity when compared with compound 6 on HEK-293 cell line. It can be inferred from the data (Table 2) that the EDB-Fn targeting conjugate was significantly effective on EDB-Fn overexpressing cells, while the conjugate has minimal effect on nonexpressing or moderately expressing cells. In conclusion, it is plausible to state that the presence of conjugating moieties on compound 11 (integrin targeting) and compound 16 (EDB-Fn targeting) significantly affects their ability to adhere and deliver the chemotherapeutic agent selectively to biomarker overexpressing cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | DU145 cell | CVCL_0105 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Breast adenocarcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
277.3 ± 14.5 nM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTS assay | ||||
| MOA of PDC |
The present study explores two different targeting peptides (TPs), RGDC (TP1), and CTVRTSAD (TP2), toward avβ1 integrin and EDB-Fn, respectively. TP1 or TP2 is covalently conjugated via disulfide bond to [C(WR)2K(WR)2]-(CBT) (6) to provide CBT conjugates (11, 16) containing an active targeting ability with increased aqueous solubility without decreasing cellular uptake. Targeting peptides may enable cCPP-CBT to target biomarker overexpressing tumor cells, and cCPP can assist in improving the physicochemical properties of CBT and delivery into the cancer cells. The conjugates in vitro stability was examined in various relevant conditions to mimic the cancerous versus normal physiological environment. Conjugates were evaluated in human plasma at two different glutathione concentrations (GSH, 1 mM and 10 mM) and two different pHs (6.5 and 7.4). The reported GSH level in noncancerous conditions is below 1 mM and approximately 10 mM at the cancer site. The in vitro antiproliferative assay using a biomarker overexpressing cell lines was compared to those without or with moderately expressing cancer cell lines. The study results demonstrate that targeting enhances the efficacy of CBT on cancer cell lines overexpressing extracellular biomarkers.
Click to Show/Hide
|
||||
| Description |
The conjugates therapeutic efficacy with targeting moieties was evaluated using prostate cancer cell lines with and without EDB-Fn overexpression (C4-2 and DU-145). Breast cancer cell lines with and without integrin v1 overexpression (MCF-7 and MDA-MB-231) were also used to evaluate antiproliferative activity. Noncancerous embryonic kidney epithelial cell line (HEK-293) was used as a control since this cell line did not express both biomarkers. All the compounds with and without targeting moieties showed comparable or lower antiproliferative activity than the CBT alone after 72 h incubation. The antiproliferative activity of compound 11 on MCF-7 (IC50 = 160.4 nM) was 1.6 times greater than on MDA-MB-231 (IC50 = 251.6 nM), showing a slight improvement in antiproliferative activity. A similar observation was found with compound 16 that showed 1.45 times higher antiproliferative activity on MCF-7 (IC50 = 162.4 nM) as compared to MDA-MB-231 cells (IC50 = 235.0 nM). Both MCF-7 and MDA-MB-231 are breast cancer cell lines. MDA-MB-231 is triple-negative breast cancer cells, and it showed higher IC50 values for CBT than MCF-7 cells. Later, the antiproliferative activity was compared between MCF-7 and the control cells (HEK-293); it was observed that compound 11 showed 3.33 times higher antiproliferative activity on MCF-7 cells (IC50 for 11 = 160.4 nM) compared to HEK-293 (IC50 for 11 = 534.5 nM), and compound 16 showed 4.6 times higher antiproliferative activity on MCF-7 (IC50 for 16 = 162.4 nM) compared to HEK-293 (IC50 for 16 = 483.3 nM). Thereafter, we wanted to assess the effectiveness of the EDB-Fn targeting moiety by comparing the antiproliferative activities on prostate cancer cell lines with and without EDB-Fn overexpression. The data showed a similar trend to that observed in breast cancer cells. For instance, compound 11 showed 3.4 times higher antiproliferative activity on C4-2 (IC50 for 11 = 181.4 nM) compared to DU-145 (IC50 for 11 = 611.2 nM). Similarly, compound 16 showed 3.7 times higher antiproliferative activity on C4-2 (IC50 for 16 = 202.2 nM) compared to DU-145 (IC50 for 16 = 751.1 nM). Compound 16 (possessing EDB-Fn targeting moiety) showed 1.1 times less antiproliferative activity compared to compound 6 (cCPP-CBT, without targeting moiety) on C4-2 cell lines, but compound 16 showed a much higher difference (3.6 times) in antiproliferative activity when compared with compound 6 on EDB-Fn moderately expressing DU-145 cell line. This data is further corroborated in EDB-Fn nonexpressing HEK-293. For instance, compound 16 showed 4.6 times less antiproliferative activity when compared with compound 6 on HEK-293 cell line. It can be inferred from the data (Table 2) that the EDB-Fn targeting conjugate was significantly effective on EDB-Fn overexpressing cells, while the conjugate has minimal effect on nonexpressing or moderately expressing cells. In conclusion, it is plausible to state that the presence of conjugating moieties on compound 11 (integrin targeting) and compound 16 (EDB-Fn targeting) significantly affects their ability to adhere and deliver the chemotherapeutic agent selectively to biomarker overexpressing cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
MAHNP-Dox conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
110.1 ± 12.7 nM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
You et al. developed a unique approach for the treatment of human epidermal growth factor receptor 2 (HER2)-positive breast cancer. A peptide fragment from the heavy chain 3 of the full-length antibody trastuzumab was obtained and termed AHNP. The 12-mer AHNP binds the extracellular domain of HER2 with high affinity and displays similar potency as trastuzumab. The peptide mimetic AHNP was then conjugated via an MMP-2 sensitive linker with doxorubicin (Dox) to form MAHNP-Dox conjugate essentially using ester/amide bonds.
Click to Show/Hide
|
||||
| Description |
Conjugate MAHNP-Dox was compared to Dox using HER2 positive breast cancer cell lines BT474 and SKBR3. Cellular toxicity analysis showed that MAHNP-Dox was more potent than free Dox, as indicated by lower IC50 values for both cell lines. The BT474 and SKBR3 cell lines had an IC50 of 746.8 ± 81.5 nM and 110.1 ± 12.7 nM for MAHNP-DOX and 2075.0 ± 368.0 nM and 172.9 ± 19.2 nM for Dox, respectively.
Click to Show/Hide
|
||||
| Experiment 2 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
746.8 ± 81.5 nM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
You et al. developed a unique approach for the treatment of human epidermal growth factor receptor 2 (HER2)-positive breast cancer. A peptide fragment from the heavy chain 3 of the full-length antibody trastuzumab was obtained and termed AHNP. The 12-mer AHNP binds the extracellular domain of HER2 with high affinity and displays similar potency as trastuzumab. The peptide mimetic AHNP was then conjugated via an MMP-2 sensitive linker with doxorubicin (Dox) to form MAHNP-Dox conjugate essentially using ester/amide bonds.
Click to Show/Hide
|
||||
| Description |
Conjugate MAHNP-Dox was compared to Dox using HER2 positive breast cancer cell lines BT474 and SKBR3. Cellular toxicity analysis showed that MAHNP-Dox was more potent than free Dox, as indicated by lower IC50 values for both cell lines. The BT474 and SKBR3 cell lines had an IC50 of 746.8 ± 81.5 nM and 110.1 ± 12.7 nM for MAHNP-DOX and 2075.0 ± 368.0 nM and 172.9 ± 19.2 nM for Dox, respectively.
Click to Show/Hide
|
||||
E1-7 doxorubicin [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Medulloblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
130 ± 1.27 nM
|
|||
| Administration Time | 72 h | ||||
| Description |
This was confirmed with E1-7 doxorubicin conjugate (4) displaying a 5-fold reduction in cytotoxicity compared to E1-3 doxorubicin conjugate (3) (IC50 values of 130 ± 1.27 nM and 25 ± 1.22 nM, respectively) and 14-fold reduction in cytotoxicity compared to free doxorubicin (5) (IC50 values of 130 ± 1.27 nM and 8.8 ± 1.31 nM, respectively) (Figure 7).
Click to Show/Hide
|
||||
| In Vitro Model | Medulloblastoma | Medulloblastoma cell | Homo sapiens | ||
TP1-cCPP-CBT [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Invasive breast carcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
160.4 ± 8.4 nM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTS assay | ||||
| MOA of PDC |
The present study explores two different targeting peptides (TPs), RGDC (TP1), and CTVRTSAD (TP2), toward avβ1 integrin and EDB-Fn, respectively. TP1 or TP2 is covalently conjugated via disulfide bond to [C(WR)2K(WR)2]-(CBT) (6) to provide CBT conjugates (11, 16) containing an active targeting ability with increased aqueous solubility without decreasing cellular uptake. Targeting peptides may enable cCPP-CBT to target biomarker overexpressing tumor cells, and cCPP can assist in improving the physicochemical properties of CBT and delivery into the cancer cells. The conjugates in vitro stability was examined in various relevant conditions to mimic the cancerous versus normal physiological environment. Conjugates were evaluated in human plasma at two different glutathione concentrations (GSH, 1 mM and 10 mM) and two different pHs (6.5 and 7.4). The reported GSH level in noncancerous conditions is below 1 mM and approximately 10 mM at the cancer site. The in vitro antiproliferative assay using a biomarker overexpressing cell lines was compared to those without or with moderately expressing cancer cell lines. The study results demonstrate that targeting enhances the efficacy of CBT on cancer cell lines overexpressing extracellular biomarkers.
Click to Show/Hide
|
||||
| Description |
The conjugates therapeutic efficacy with targeting moieties was evaluated using prostate cancer cell lines with and without EDB-Fn overexpression (C4-2 and DU-145). Breast cancer cell lines with and without integrin v1 overexpression (MCF-7 and MDA-MB-231) were also used to evaluate antiproliferative activity. Noncancerous embryonic kidney epithelial cell line (HEK-293) was used as a control since this cell line did not express both biomarkers. All the compounds with and without targeting moieties showed comparable or lower antiproliferative activity than the CBT alone after 72 h incubation. The antiproliferative activity of compound 11 on MCF-7 (IC50 = 160.4 nM) was 1.6 times greater than on MDA-MB-231 (IC50 = 251.6 nM), showing a slight improvement in antiproliferative activity. A similar observation was found with compound 16 that showed 1.45 times higher antiproliferative activity on MCF-7 (IC50 = 162.4 nM) as compared to MDA-MB-231 cells (IC50 = 235.0 nM). Both MCF-7 and MDA-MB-231 are breast cancer cell lines. MDA-MB-231 is triple-negative breast cancer cells, and it showed higher IC50 values for CBT than MCF-7 cells. Later, the antiproliferative activity was compared between MCF-7 and the control cells (HEK-293); it was observed that compound 11 showed 3.33 times higher antiproliferative activity on MCF-7 cells (IC50 for 11 = 160.4 nM) compared to HEK-293 (IC50 for 11 = 534.5 nM), and compound 16 showed 4.6 times higher antiproliferative activity on MCF-7 (IC50 for 16 = 162.4 nM) compared to HEK-293 (IC50 for 16 = 483.3 nM). Thereafter, we wanted to assess the effectiveness of the EDB-Fn targeting moiety by comparing the antiproliferative activities on prostate cancer cell lines with and without EDB-Fn overexpression. The data showed a similar trend to that observed in breast cancer cells. For instance, compound 11 showed 3.4 times higher antiproliferative activity on C4-2 (IC50 for 11 = 181.4 nM) compared to DU-145 (IC50 for 11 = 611.2 nM). Similarly, compound 16 showed 3.7 times higher antiproliferative activity on C4-2 (IC50 for 16 = 202.2 nM) compared to DU-145 (IC50 for 16 = 751.1 nM). Compound 16 (possessing EDB-Fn targeting moiety) showed 1.1 times less antiproliferative activity compared to compound 6 (cCPP-CBT, without targeting moiety) on C4-2 cell lines, but compound 16 showed a much higher difference (3.6 times) in antiproliferative activity when compared with compound 6 on EDB-Fn moderately expressing DU-145 cell line. This data is further corroborated in EDB-Fn nonexpressing HEK-293. For instance, compound 16 showed 4.6 times less antiproliferative activity when compared with compound 6 on HEK-293 cell line. It can be inferred from the data (Table 2) that the EDB-Fn targeting conjugate was significantly effective on EDB-Fn overexpressing cells, while the conjugate has minimal effect on nonexpressing or moderately expressing cells. In conclusion, it is plausible to state that the presence of conjugating moieties on compound 11 (integrin targeting) and compound 16 (EDB-Fn targeting) significantly affects their ability to adhere and deliver the chemotherapeutic agent selectively to biomarker overexpressing cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Half life period | 1.013 h | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Prostate carcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
181.4 ± 8.7 nM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTS assay | ||||
| MOA of PDC |
The present study explores two different targeting peptides (TPs), RGDC (TP1), and CTVRTSAD (TP2), toward avβ1 integrin and EDB-Fn, respectively. TP1 or TP2 is covalently conjugated via disulfide bond to [C(WR)2K(WR)2]-(CBT) (6) to provide CBT conjugates (11, 16) containing an active targeting ability with increased aqueous solubility without decreasing cellular uptake. Targeting peptides may enable cCPP-CBT to target biomarker overexpressing tumor cells, and cCPP can assist in improving the physicochemical properties of CBT and delivery into the cancer cells. The conjugates in vitro stability was examined in various relevant conditions to mimic the cancerous versus normal physiological environment. Conjugates were evaluated in human plasma at two different glutathione concentrations (GSH, 1 mM and 10 mM) and two different pHs (6.5 and 7.4). The reported GSH level in noncancerous conditions is below 1 mM and approximately 10 mM at the cancer site. The in vitro antiproliferative assay using a biomarker overexpressing cell lines was compared to those without or with moderately expressing cancer cell lines. The study results demonstrate that targeting enhances the efficacy of CBT on cancer cell lines overexpressing extracellular biomarkers.
Click to Show/Hide
|
||||
| Description |
The conjugates therapeutic efficacy with targeting moieties was evaluated using prostate cancer cell lines with and without EDB-Fn overexpression (C4-2 and DU-145). Breast cancer cell lines with and without integrin v1 overexpression (MCF-7 and MDA-MB-231) were also used to evaluate antiproliferative activity. Noncancerous embryonic kidney epithelial cell line (HEK-293) was used as a control since this cell line did not express both biomarkers. All the compounds with and without targeting moieties showed comparable or lower antiproliferative activity than the CBT alone after 72 h incubation. The antiproliferative activity of compound 11 on MCF-7 (IC50 = 160.4 nM) was 1.6 times greater than on MDA-MB-231 (IC50 = 251.6 nM), showing a slight improvement in antiproliferative activity. A similar observation was found with compound 16 that showed 1.45 times higher antiproliferative activity on MCF-7 (IC50 = 162.4 nM) as compared to MDA-MB-231 cells (IC50 = 235.0 nM). Both MCF-7 and MDA-MB-231 are breast cancer cell lines. MDA-MB-231 is triple-negative breast cancer cells, and it showed higher IC50 values for CBT than MCF-7 cells. Later, the antiproliferative activity was compared between MCF-7 and the control cells (HEK-293); it was observed that compound 11 showed 3.33 times higher antiproliferative activity on MCF-7 cells (IC50 for 11 = 160.4 nM) compared to HEK-293 (IC50 for 11 = 534.5 nM), and compound 16 showed 4.6 times higher antiproliferative activity on MCF-7 (IC50 for 16 = 162.4 nM) compared to HEK-293 (IC50 for 16 = 483.3 nM). Thereafter, we wanted to assess the effectiveness of the EDB-Fn targeting moiety by comparing the antiproliferative activities on prostate cancer cell lines with and without EDB-Fn overexpression. The data showed a similar trend to that observed in breast cancer cells. For instance, compound 11 showed 3.4 times higher antiproliferative activity on C4-2 (IC50 for 11 = 181.4 nM) compared to DU-145 (IC50 for 11 = 611.2 nM). Similarly, compound 16 showed 3.7 times higher antiproliferative activity on C4-2 (IC50 for 16 = 202.2 nM) compared to DU-145 (IC50 for 16 = 751.1 nM). Compound 16 (possessing EDB-Fn targeting moiety) showed 1.1 times less antiproliferative activity compared to compound 6 (cCPP-CBT, without targeting moiety) on C4-2 cell lines, but compound 16 showed a much higher difference (3.6 times) in antiproliferative activity when compared with compound 6 on EDB-Fn moderately expressing DU-145 cell line. This data is further corroborated in EDB-Fn nonexpressing HEK-293. For instance, compound 16 showed 4.6 times less antiproliferative activity when compared with compound 6 on HEK-293 cell line. It can be inferred from the data (Table 2) that the EDB-Fn targeting conjugate was significantly effective on EDB-Fn overexpressing cells, while the conjugate has minimal effect on nonexpressing or moderately expressing cells. In conclusion, it is plausible to state that the presence of conjugating moieties on compound 11 (integrin targeting) and compound 16 (EDB-Fn targeting) significantly affects their ability to adhere and deliver the chemotherapeutic agent selectively to biomarker overexpressing cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | LNCaP C4-2 cell | CVCL_4782 | ||
| Half life period | 1.013 h | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Breast adenocarcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
251.6 ± 17.6 nM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTS assay | ||||
| MOA of PDC |
The present study explores two different targeting peptides (TPs), RGDC (TP1), and CTVRTSAD (TP2), toward avβ1 integrin and EDB-Fn, respectively. TP1 or TP2 is covalently conjugated via disulfide bond to [C(WR)2K(WR)2]-(CBT) (6) to provide CBT conjugates (11, 16) containing an active targeting ability with increased aqueous solubility without decreasing cellular uptake. Targeting peptides may enable cCPP-CBT to target biomarker overexpressing tumor cells, and cCPP can assist in improving the physicochemical properties of CBT and delivery into the cancer cells. The conjugates in vitro stability was examined in various relevant conditions to mimic the cancerous versus normal physiological environment. Conjugates were evaluated in human plasma at two different glutathione concentrations (GSH, 1 mM and 10 mM) and two different pHs (6.5 and 7.4). The reported GSH level in noncancerous conditions is below 1 mM and approximately 10 mM at the cancer site. The in vitro antiproliferative assay using a biomarker overexpressing cell lines was compared to those without or with moderately expressing cancer cell lines. The study results demonstrate that targeting enhances the efficacy of CBT on cancer cell lines overexpressing extracellular biomarkers.
Click to Show/Hide
|
||||
| Description |
The conjugates therapeutic efficacy with targeting moieties was evaluated using prostate cancer cell lines with and without EDB-Fn overexpression (C4-2 and DU-145). Breast cancer cell lines with and without integrin v1 overexpression (MCF-7 and MDA-MB-231) were also used to evaluate antiproliferative activity. Noncancerous embryonic kidney epithelial cell line (HEK-293) was used as a control since this cell line did not express both biomarkers. All the compounds with and without targeting moieties showed comparable or lower antiproliferative activity than the CBT alone after 72 h incubation. The antiproliferative activity of compound 11 on MCF-7 (IC50 = 160.4 nM) was 1.6 times greater than on MDA-MB-231 (IC50 = 251.6 nM), showing a slight improvement in antiproliferative activity. A similar observation was found with compound 16 that showed 1.45 times higher antiproliferative activity on MCF-7 (IC50 = 162.4 nM) as compared to MDA-MB-231 cells (IC50 = 235.0 nM). Both MCF-7 and MDA-MB-231 are breast cancer cell lines. MDA-MB-231 is triple-negative breast cancer cells, and it showed higher IC50 values for CBT than MCF-7 cells. Later, the antiproliferative activity was compared between MCF-7 and the control cells (HEK-293); it was observed that compound 11 showed 3.33 times higher antiproliferative activity on MCF-7 cells (IC50 for 11 = 160.4 nM) compared to HEK-293 (IC50 for 11 = 534.5 nM), and compound 16 showed 4.6 times higher antiproliferative activity on MCF-7 (IC50 for 16 = 162.4 nM) compared to HEK-293 (IC50 for 16 = 483.3 nM). Thereafter, we wanted to assess the effectiveness of the EDB-Fn targeting moiety by comparing the antiproliferative activities on prostate cancer cell lines with and without EDB-Fn overexpression. The data showed a similar trend to that observed in breast cancer cells. For instance, compound 11 showed 3.4 times higher antiproliferative activity on C4-2 (IC50 for 11 = 181.4 nM) compared to DU-145 (IC50 for 11 = 611.2 nM). Similarly, compound 16 showed 3.7 times higher antiproliferative activity on C4-2 (IC50 for 16 = 202.2 nM) compared to DU-145 (IC50 for 16 = 751.1 nM). Compound 16 (possessing EDB-Fn targeting moiety) showed 1.1 times less antiproliferative activity compared to compound 6 (cCPP-CBT, without targeting moiety) on C4-2 cell lines, but compound 16 showed a much higher difference (3.6 times) in antiproliferative activity when compared with compound 6 on EDB-Fn moderately expressing DU-145 cell line. This data is further corroborated in EDB-Fn nonexpressing HEK-293. For instance, compound 16 showed 4.6 times less antiproliferative activity when compared with compound 6 on HEK-293 cell line. It can be inferred from the data (Table 2) that the EDB-Fn targeting conjugate was significantly effective on EDB-Fn overexpressing cells, while the conjugate has minimal effect on nonexpressing or moderately expressing cells. In conclusion, it is plausible to state that the presence of conjugating moieties on compound 11 (integrin targeting) and compound 16 (EDB-Fn targeting) significantly affects their ability to adhere and deliver the chemotherapeutic agent selectively to biomarker overexpressing cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Half life period | 1.013 h | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [6] | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
534.5 ± 23.3 nM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTS assay | ||||
| MOA of PDC |
The present study explores two different targeting peptides (TPs), RGDC (TP1), and CTVRTSAD (TP2), toward avβ1 integrin and EDB-Fn, respectively. TP1 or TP2 is covalently conjugated via disulfide bond to [C(WR)2K(WR)2]-(CBT) (6) to provide CBT conjugates (11, 16) containing an active targeting ability with increased aqueous solubility without decreasing cellular uptake. Targeting peptides may enable cCPP-CBT to target biomarker overexpressing tumor cells, and cCPP can assist in improving the physicochemical properties of CBT and delivery into the cancer cells. The conjugates in vitro stability was examined in various relevant conditions to mimic the cancerous versus normal physiological environment. Conjugates were evaluated in human plasma at two different glutathione concentrations (GSH, 1 mM and 10 mM) and two different pHs (6.5 and 7.4). The reported GSH level in noncancerous conditions is below 1 mM and approximately 10 mM at the cancer site. The in vitro antiproliferative assay using a biomarker overexpressing cell lines was compared to those without or with moderately expressing cancer cell lines. The study results demonstrate that targeting enhances the efficacy of CBT on cancer cell lines overexpressing extracellular biomarkers.
Click to Show/Hide
|
||||
| Description |
The conjugates therapeutic efficacy with targeting moieties was evaluated using prostate cancer cell lines with and without EDB-Fn overexpression (C4-2 and DU-145). Breast cancer cell lines with and without integrin v1 overexpression (MCF-7 and MDA-MB-231) were also used to evaluate antiproliferative activity. Noncancerous embryonic kidney epithelial cell line (HEK-293) was used as a control since this cell line did not express both biomarkers. All the compounds with and without targeting moieties showed comparable or lower antiproliferative activity than the CBT alone after 72 h incubation. The antiproliferative activity of compound 11 on MCF-7 (IC50 = 160.4 nM) was 1.6 times greater than on MDA-MB-231 (IC50 = 251.6 nM), showing a slight improvement in antiproliferative activity. A similar observation was found with compound 16 that showed 1.45 times higher antiproliferative activity on MCF-7 (IC50 = 162.4 nM) as compared to MDA-MB-231 cells (IC50 = 235.0 nM). Both MCF-7 and MDA-MB-231 are breast cancer cell lines. MDA-MB-231 is triple-negative breast cancer cells, and it showed higher IC50 values for CBT than MCF-7 cells. Later, the antiproliferative activity was compared between MCF-7 and the control cells (HEK-293); it was observed that compound 11 showed 3.33 times higher antiproliferative activity on MCF-7 cells (IC50 for 11 = 160.4 nM) compared to HEK-293 (IC50 for 11 = 534.5 nM), and compound 16 showed 4.6 times higher antiproliferative activity on MCF-7 (IC50 for 16 = 162.4 nM) compared to HEK-293 (IC50 for 16 = 483.3 nM). Thereafter, we wanted to assess the effectiveness of the EDB-Fn targeting moiety by comparing the antiproliferative activities on prostate cancer cell lines with and without EDB-Fn overexpression. The data showed a similar trend to that observed in breast cancer cells. For instance, compound 11 showed 3.4 times higher antiproliferative activity on C4-2 (IC50 for 11 = 181.4 nM) compared to DU-145 (IC50 for 11 = 611.2 nM). Similarly, compound 16 showed 3.7 times higher antiproliferative activity on C4-2 (IC50 for 16 = 202.2 nM) compared to DU-145 (IC50 for 16 = 751.1 nM). Compound 16 (possessing EDB-Fn targeting moiety) showed 1.1 times less antiproliferative activity compared to compound 6 (cCPP-CBT, without targeting moiety) on C4-2 cell lines, but compound 16 showed a much higher difference (3.6 times) in antiproliferative activity when compared with compound 6 on EDB-Fn moderately expressing DU-145 cell line. This data is further corroborated in EDB-Fn nonexpressing HEK-293. For instance, compound 16 showed 4.6 times less antiproliferative activity when compared with compound 6 on HEK-293 cell line. It can be inferred from the data (Table 2) that the EDB-Fn targeting conjugate was significantly effective on EDB-Fn overexpressing cells, while the conjugate has minimal effect on nonexpressing or moderately expressing cells. In conclusion, it is plausible to state that the presence of conjugating moieties on compound 11 (integrin targeting) and compound 16 (EDB-Fn targeting) significantly affects their ability to adhere and deliver the chemotherapeutic agent selectively to biomarker overexpressing cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | HEK293 cell | CVCL_0045 | ||
| Half life period | 1.013 h | ||||
| Experiment 5 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Prostate carcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
611.2 ± 18.4 nM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTS assay | ||||
| MOA of PDC |
The present study explores two different targeting peptides (TPs), RGDC (TP1), and CTVRTSAD (TP2), toward avβ1 integrin and EDB-Fn, respectively. TP1 or TP2 is covalently conjugated via disulfide bond to [C(WR)2K(WR)2]-(CBT) (6) to provide CBT conjugates (11, 16) containing an active targeting ability with increased aqueous solubility without decreasing cellular uptake. Targeting peptides may enable cCPP-CBT to target biomarker overexpressing tumor cells, and cCPP can assist in improving the physicochemical properties of CBT and delivery into the cancer cells. The conjugates in vitro stability was examined in various relevant conditions to mimic the cancerous versus normal physiological environment. Conjugates were evaluated in human plasma at two different glutathione concentrations (GSH, 1 mM and 10 mM) and two different pHs (6.5 and 7.4). The reported GSH level in noncancerous conditions is below 1 mM and approximately 10 mM at the cancer site. The in vitro antiproliferative assay using a biomarker overexpressing cell lines was compared to those without or with moderately expressing cancer cell lines. The study results demonstrate that targeting enhances the efficacy of CBT on cancer cell lines overexpressing extracellular biomarkers.
Click to Show/Hide
|
||||
| Description |
The conjugates therapeutic efficacy with targeting moieties was evaluated using prostate cancer cell lines with and without EDB-Fn overexpression (C4-2 and DU-145). Breast cancer cell lines with and without integrin v1 overexpression (MCF-7 and MDA-MB-231) were also used to evaluate antiproliferative activity. Noncancerous embryonic kidney epithelial cell line (HEK-293) was used as a control since this cell line did not express both biomarkers. All the compounds with and without targeting moieties showed comparable or lower antiproliferative activity than the CBT alone after 72 h incubation. The antiproliferative activity of compound 11 on MCF-7 (IC50 = 160.4 nM) was 1.6 times greater than on MDA-MB-231 (IC50 = 251.6 nM), showing a slight improvement in antiproliferative activity. A similar observation was found with compound 16 that showed 1.45 times higher antiproliferative activity on MCF-7 (IC50 = 162.4 nM) as compared to MDA-MB-231 cells (IC50 = 235.0 nM). Both MCF-7 and MDA-MB-231 are breast cancer cell lines. MDA-MB-231 is triple-negative breast cancer cells, and it showed higher IC50 values for CBT than MCF-7 cells. Later, the antiproliferative activity was compared between MCF-7 and the control cells (HEK-293); it was observed that compound 11 showed 3.33 times higher antiproliferative activity on MCF-7 cells (IC50 for 11 = 160.4 nM) compared to HEK-293 (IC50 for 11 = 534.5 nM), and compound 16 showed 4.6 times higher antiproliferative activity on MCF-7 (IC50 for 16 = 162.4 nM) compared to HEK-293 (IC50 for 16 = 483.3 nM). Thereafter, we wanted to assess the effectiveness of the EDB-Fn targeting moiety by comparing the antiproliferative activities on prostate cancer cell lines with and without EDB-Fn overexpression. The data showed a similar trend to that observed in breast cancer cells. For instance, compound 11 showed 3.4 times higher antiproliferative activity on C4-2 (IC50 for 11 = 181.4 nM) compared to DU-145 (IC50 for 11 = 611.2 nM). Similarly, compound 16 showed 3.7 times higher antiproliferative activity on C4-2 (IC50 for 16 = 202.2 nM) compared to DU-145 (IC50 for 16 = 751.1 nM). Compound 16 (possessing EDB-Fn targeting moiety) showed 1.1 times less antiproliferative activity compared to compound 6 (cCPP-CBT, without targeting moiety) on C4-2 cell lines, but compound 16 showed a much higher difference (3.6 times) in antiproliferative activity when compared with compound 6 on EDB-Fn moderately expressing DU-145 cell line. This data is further corroborated in EDB-Fn nonexpressing HEK-293. For instance, compound 16 showed 4.6 times less antiproliferative activity when compared with compound 6 on HEK-293 cell line. It can be inferred from the data (Table 2) that the EDB-Fn targeting conjugate was significantly effective on EDB-Fn overexpressing cells, while the conjugate has minimal effect on nonexpressing or moderately expressing cells. In conclusion, it is plausible to state that the presence of conjugating moieties on compound 11 (integrin targeting) and compound 16 (EDB-Fn targeting) significantly affects their ability to adhere and deliver the chemotherapeutic agent selectively to biomarker overexpressing cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | DU145 cell | CVCL_0105 | ||
| Half life period | 1.013 h | ||||
TP2-cCPP-CBT [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Invasive breast carcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
162.4 ± 3.2 nM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTS assay | ||||
| MOA of PDC |
The present study explores two different targeting peptides (TPs), RGDC (TP1), and CTVRTSAD (TP2), toward avβ1 integrin and EDB-Fn, respectively. TP1 or TP2 is covalently conjugated via disulfide bond to [C(WR)2K(WR)2]-(CBT) (6) to provide CBT conjugates (11, 16) containing an active targeting ability with increased aqueous solubility without decreasing cellular uptake. Targeting peptides may enable cCPP-CBT to target biomarker overexpressing tumor cells, and cCPP can assist in improving the physicochemical properties of CBT and delivery into the cancer cells. The conjugates in vitro stability was examined in various relevant conditions to mimic the cancerous versus normal physiological environment. Conjugates were evaluated in human plasma at two different glutathione concentrations (GSH, 1 mM and 10 mM) and two different pHs (6.5 and 7.4). The reported GSH level in noncancerous conditions is below 1 mM and approximately 10 mM at the cancer site. The in vitro antiproliferative assay using a biomarker overexpressing cell lines was compared to those without or with moderately expressing cancer cell lines. The study results demonstrate that targeting enhances the efficacy of CBT on cancer cell lines overexpressing extracellular biomarkers.
Click to Show/Hide
|
||||
| Description |
The conjugates therapeutic efficacy with targeting moieties was evaluated using prostate cancer cell lines with and without EDB-Fn overexpression (C4-2 and DU-145). Breast cancer cell lines with and without integrin v1 overexpression (MCF-7 and MDA-MB-231) were also used to evaluate antiproliferative activity. Noncancerous embryonic kidney epithelial cell line (HEK-293) was used as a control since this cell line did not express both biomarkers. All the compounds with and without targeting moieties showed comparable or lower antiproliferative activity than the CBT alone after 72 h incubation. The antiproliferative activity of compound 11 on MCF-7 (IC50 = 160.4 nM) was 1.6 times greater than on MDA-MB-231 (IC50 = 251.6 nM), showing a slight improvement in antiproliferative activity. A similar observation was found with compound 16 that showed 1.45 times higher antiproliferative activity on MCF-7 (IC50 = 162.4 nM) as compared to MDA-MB-231 cells (IC50 = 235.0 nM). Both MCF-7 and MDA-MB-231 are breast cancer cell lines. MDA-MB-231 is triple-negative breast cancer cells, and it showed higher IC50 values for CBT than MCF-7 cells. Later, the antiproliferative activity was compared between MCF-7 and the control cells (HEK-293); it was observed that compound 11 showed 3.33 times higher antiproliferative activity on MCF-7 cells (IC50 for 11 = 160.4 nM) compared to HEK-293 (IC50 for 11 = 534.5 nM), and compound 16 showed 4.6 times higher antiproliferative activity on MCF-7 (IC50 for 16 = 162.4 nM) compared to HEK-293 (IC50 for 16 = 483.3 nM). Thereafter, we wanted to assess the effectiveness of the EDB-Fn targeting moiety by comparing the antiproliferative activities on prostate cancer cell lines with and without EDB-Fn overexpression. The data showed a similar trend to that observed in breast cancer cells. For instance, compound 11 showed 3.4 times higher antiproliferative activity on C4-2 (IC50 for 11 = 181.4 nM) compared to DU-145 (IC50 for 11 = 611.2 nM). Similarly, compound 16 showed 3.7 times higher antiproliferative activity on C4-2 (IC50 for 16 = 202.2 nM) compared to DU-145 (IC50 for 16 = 751.1 nM). Compound 16 (possessing EDB-Fn targeting moiety) showed 1.1 times less antiproliferative activity compared to compound 6 (cCPP-CBT, without targeting moiety) on C4-2 cell lines, but compound 16 showed a much higher difference (3.6 times) in antiproliferative activity when compared with compound 6 on EDB-Fn moderately expressing DU-145 cell line. This data is further corroborated in EDB-Fn nonexpressing HEK-293. For instance, compound 16 showed 4.6 times less antiproliferative activity when compared with compound 6 on HEK-293 cell line. It can be inferred from the data (Table 2) that the EDB-Fn targeting conjugate was significantly effective on EDB-Fn overexpressing cells, while the conjugate has minimal effect on nonexpressing or moderately expressing cells. In conclusion, it is plausible to state that the presence of conjugating moieties on compound 11 (integrin targeting) and compound 16 (EDB-Fn targeting) significantly affects their ability to adhere and deliver the chemotherapeutic agent selectively to biomarker overexpressing cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Prostate carcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
202.2 ± 7.9 nM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTS assay | ||||
| MOA of PDC |
The present study explores two different targeting peptides (TPs), RGDC (TP1), and CTVRTSAD (TP2), toward avβ1 integrin and EDB-Fn, respectively. TP1 or TP2 is covalently conjugated via disulfide bond to [C(WR)2K(WR)2]-(CBT) (6) to provide CBT conjugates (11, 16) containing an active targeting ability with increased aqueous solubility without decreasing cellular uptake. Targeting peptides may enable cCPP-CBT to target biomarker overexpressing tumor cells, and cCPP can assist in improving the physicochemical properties of CBT and delivery into the cancer cells. The conjugates in vitro stability was examined in various relevant conditions to mimic the cancerous versus normal physiological environment. Conjugates were evaluated in human plasma at two different glutathione concentrations (GSH, 1 mM and 10 mM) and two different pHs (6.5 and 7.4). The reported GSH level in noncancerous conditions is below 1 mM and approximately 10 mM at the cancer site. The in vitro antiproliferative assay using a biomarker overexpressing cell lines was compared to those without or with moderately expressing cancer cell lines. The study results demonstrate that targeting enhances the efficacy of CBT on cancer cell lines overexpressing extracellular biomarkers.
Click to Show/Hide
|
||||
| Description |
The conjugates therapeutic efficacy with targeting moieties was evaluated using prostate cancer cell lines with and without EDB-Fn overexpression (C4-2 and DU-145). Breast cancer cell lines with and without integrin v1 overexpression (MCF-7 and MDA-MB-231) were also used to evaluate antiproliferative activity. Noncancerous embryonic kidney epithelial cell line (HEK-293) was used as a control since this cell line did not express both biomarkers. All the compounds with and without targeting moieties showed comparable or lower antiproliferative activity than the CBT alone after 72 h incubation. The antiproliferative activity of compound 11 on MCF-7 (IC50 = 160.4 nM) was 1.6 times greater than on MDA-MB-231 (IC50 = 251.6 nM), showing a slight improvement in antiproliferative activity. A similar observation was found with compound 16 that showed 1.45 times higher antiproliferative activity on MCF-7 (IC50 = 162.4 nM) as compared to MDA-MB-231 cells (IC50 = 235.0 nM). Both MCF-7 and MDA-MB-231 are breast cancer cell lines. MDA-MB-231 is triple-negative breast cancer cells, and it showed higher IC50 values for CBT than MCF-7 cells. Later, the antiproliferative activity was compared between MCF-7 and the control cells (HEK-293); it was observed that compound 11 showed 3.33 times higher antiproliferative activity on MCF-7 cells (IC50 for 11 = 160.4 nM) compared to HEK-293 (IC50 for 11 = 534.5 nM), and compound 16 showed 4.6 times higher antiproliferative activity on MCF-7 (IC50 for 16 = 162.4 nM) compared to HEK-293 (IC50 for 16 = 483.3 nM). Thereafter, we wanted to assess the effectiveness of the EDB-Fn targeting moiety by comparing the antiproliferative activities on prostate cancer cell lines with and without EDB-Fn overexpression. The data showed a similar trend to that observed in breast cancer cells. For instance, compound 11 showed 3.4 times higher antiproliferative activity on C4-2 (IC50 for 11 = 181.4 nM) compared to DU-145 (IC50 for 11 = 611.2 nM). Similarly, compound 16 showed 3.7 times higher antiproliferative activity on C4-2 (IC50 for 16 = 202.2 nM) compared to DU-145 (IC50 for 16 = 751.1 nM). Compound 16 (possessing EDB-Fn targeting moiety) showed 1.1 times less antiproliferative activity compared to compound 6 (cCPP-CBT, without targeting moiety) on C4-2 cell lines, but compound 16 showed a much higher difference (3.6 times) in antiproliferative activity when compared with compound 6 on EDB-Fn moderately expressing DU-145 cell line. This data is further corroborated in EDB-Fn nonexpressing HEK-293. For instance, compound 16 showed 4.6 times less antiproliferative activity when compared with compound 6 on HEK-293 cell line. It can be inferred from the data (Table 2) that the EDB-Fn targeting conjugate was significantly effective on EDB-Fn overexpressing cells, while the conjugate has minimal effect on nonexpressing or moderately expressing cells. In conclusion, it is plausible to state that the presence of conjugating moieties on compound 11 (integrin targeting) and compound 16 (EDB-Fn targeting) significantly affects their ability to adhere and deliver the chemotherapeutic agent selectively to biomarker overexpressing cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | LNCaP C4-2 cell | CVCL_4782 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Breast adenocarcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
235 ± 11.4 nM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTS assay | ||||
| MOA of PDC |
The present study explores two different targeting peptides (TPs), RGDC (TP1), and CTVRTSAD (TP2), toward avβ1 integrin and EDB-Fn, respectively. TP1 or TP2 is covalently conjugated via disulfide bond to [C(WR)2K(WR)2]-(CBT) (6) to provide CBT conjugates (11, 16) containing an active targeting ability with increased aqueous solubility without decreasing cellular uptake. Targeting peptides may enable cCPP-CBT to target biomarker overexpressing tumor cells, and cCPP can assist in improving the physicochemical properties of CBT and delivery into the cancer cells. The conjugates in vitro stability was examined in various relevant conditions to mimic the cancerous versus normal physiological environment. Conjugates were evaluated in human plasma at two different glutathione concentrations (GSH, 1 mM and 10 mM) and two different pHs (6.5 and 7.4). The reported GSH level in noncancerous conditions is below 1 mM and approximately 10 mM at the cancer site. The in vitro antiproliferative assay using a biomarker overexpressing cell lines was compared to those without or with moderately expressing cancer cell lines. The study results demonstrate that targeting enhances the efficacy of CBT on cancer cell lines overexpressing extracellular biomarkers.
Click to Show/Hide
|
||||
| Description |
The conjugates therapeutic efficacy with targeting moieties was evaluated using prostate cancer cell lines with and without EDB-Fn overexpression (C4-2 and DU-145). Breast cancer cell lines with and without integrin v1 overexpression (MCF-7 and MDA-MB-231) were also used to evaluate antiproliferative activity. Noncancerous embryonic kidney epithelial cell line (HEK-293) was used as a control since this cell line did not express both biomarkers. All the compounds with and without targeting moieties showed comparable or lower antiproliferative activity than the CBT alone after 72 h incubation. The antiproliferative activity of compound 11 on MCF-7 (IC50 = 160.4 nM) was 1.6 times greater than on MDA-MB-231 (IC50 = 251.6 nM), showing a slight improvement in antiproliferative activity. A similar observation was found with compound 16 that showed 1.45 times higher antiproliferative activity on MCF-7 (IC50 = 162.4 nM) as compared to MDA-MB-231 cells (IC50 = 235.0 nM). Both MCF-7 and MDA-MB-231 are breast cancer cell lines. MDA-MB-231 is triple-negative breast cancer cells, and it showed higher IC50 values for CBT than MCF-7 cells. Later, the antiproliferative activity was compared between MCF-7 and the control cells (HEK-293); it was observed that compound 11 showed 3.33 times higher antiproliferative activity on MCF-7 cells (IC50 for 11 = 160.4 nM) compared to HEK-293 (IC50 for 11 = 534.5 nM), and compound 16 showed 4.6 times higher antiproliferative activity on MCF-7 (IC50 for 16 = 162.4 nM) compared to HEK-293 (IC50 for 16 = 483.3 nM). Thereafter, we wanted to assess the effectiveness of the EDB-Fn targeting moiety by comparing the antiproliferative activities on prostate cancer cell lines with and without EDB-Fn overexpression. The data showed a similar trend to that observed in breast cancer cells. For instance, compound 11 showed 3.4 times higher antiproliferative activity on C4-2 (IC50 for 11 = 181.4 nM) compared to DU-145 (IC50 for 11 = 611.2 nM). Similarly, compound 16 showed 3.7 times higher antiproliferative activity on C4-2 (IC50 for 16 = 202.2 nM) compared to DU-145 (IC50 for 16 = 751.1 nM). Compound 16 (possessing EDB-Fn targeting moiety) showed 1.1 times less antiproliferative activity compared to compound 6 (cCPP-CBT, without targeting moiety) on C4-2 cell lines, but compound 16 showed a much higher difference (3.6 times) in antiproliferative activity when compared with compound 6 on EDB-Fn moderately expressing DU-145 cell line. This data is further corroborated in EDB-Fn nonexpressing HEK-293. For instance, compound 16 showed 4.6 times less antiproliferative activity when compared with compound 6 on HEK-293 cell line. It can be inferred from the data (Table 2) that the EDB-Fn targeting conjugate was significantly effective on EDB-Fn overexpressing cells, while the conjugate has minimal effect on nonexpressing or moderately expressing cells. In conclusion, it is plausible to state that the presence of conjugating moieties on compound 11 (integrin targeting) and compound 16 (EDB-Fn targeting) significantly affects their ability to adhere and deliver the chemotherapeutic agent selectively to biomarker overexpressing cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [6] | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
483.3 ± 17.2 nM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTS assay | ||||
| MOA of PDC |
The present study explores two different targeting peptides (TPs), RGDC (TP1), and CTVRTSAD (TP2), toward avβ1 integrin and EDB-Fn, respectively. TP1 or TP2 is covalently conjugated via disulfide bond to [C(WR)2K(WR)2]-(CBT) (6) to provide CBT conjugates (11, 16) containing an active targeting ability with increased aqueous solubility without decreasing cellular uptake. Targeting peptides may enable cCPP-CBT to target biomarker overexpressing tumor cells, and cCPP can assist in improving the physicochemical properties of CBT and delivery into the cancer cells. The conjugates in vitro stability was examined in various relevant conditions to mimic the cancerous versus normal physiological environment. Conjugates were evaluated in human plasma at two different glutathione concentrations (GSH, 1 mM and 10 mM) and two different pHs (6.5 and 7.4). The reported GSH level in noncancerous conditions is below 1 mM and approximately 10 mM at the cancer site. The in vitro antiproliferative assay using a biomarker overexpressing cell lines was compared to those without or with moderately expressing cancer cell lines. The study results demonstrate that targeting enhances the efficacy of CBT on cancer cell lines overexpressing extracellular biomarkers.
Click to Show/Hide
|
||||
| Description |
The conjugates therapeutic efficacy with targeting moieties was evaluated using prostate cancer cell lines with and without EDB-Fn overexpression (C4-2 and DU-145). Breast cancer cell lines with and without integrin v1 overexpression (MCF-7 and MDA-MB-231) were also used to evaluate antiproliferative activity. Noncancerous embryonic kidney epithelial cell line (HEK-293) was used as a control since this cell line did not express both biomarkers. All the compounds with and without targeting moieties showed comparable or lower antiproliferative activity than the CBT alone after 72 h incubation. The antiproliferative activity of compound 11 on MCF-7 (IC50 = 160.4 nM) was 1.6 times greater than on MDA-MB-231 (IC50 = 251.6 nM), showing a slight improvement in antiproliferative activity. A similar observation was found with compound 16 that showed 1.45 times higher antiproliferative activity on MCF-7 (IC50 = 162.4 nM) as compared to MDA-MB-231 cells (IC50 = 235.0 nM). Both MCF-7 and MDA-MB-231 are breast cancer cell lines. MDA-MB-231 is triple-negative breast cancer cells, and it showed higher IC50 values for CBT than MCF-7 cells. Later, the antiproliferative activity was compared between MCF-7 and the control cells (HEK-293); it was observed that compound 11 showed 3.33 times higher antiproliferative activity on MCF-7 cells (IC50 for 11 = 160.4 nM) compared to HEK-293 (IC50 for 11 = 534.5 nM), and compound 16 showed 4.6 times higher antiproliferative activity on MCF-7 (IC50 for 16 = 162.4 nM) compared to HEK-293 (IC50 for 16 = 483.3 nM). Thereafter, we wanted to assess the effectiveness of the EDB-Fn targeting moiety by comparing the antiproliferative activities on prostate cancer cell lines with and without EDB-Fn overexpression. The data showed a similar trend to that observed in breast cancer cells. For instance, compound 11 showed 3.4 times higher antiproliferative activity on C4-2 (IC50 for 11 = 181.4 nM) compared to DU-145 (IC50 for 11 = 611.2 nM). Similarly, compound 16 showed 3.7 times higher antiproliferative activity on C4-2 (IC50 for 16 = 202.2 nM) compared to DU-145 (IC50 for 16 = 751.1 nM). Compound 16 (possessing EDB-Fn targeting moiety) showed 1.1 times less antiproliferative activity compared to compound 6 (cCPP-CBT, without targeting moiety) on C4-2 cell lines, but compound 16 showed a much higher difference (3.6 times) in antiproliferative activity when compared with compound 6 on EDB-Fn moderately expressing DU-145 cell line. This data is further corroborated in EDB-Fn nonexpressing HEK-293. For instance, compound 16 showed 4.6 times less antiproliferative activity when compared with compound 6 on HEK-293 cell line. It can be inferred from the data (Table 2) that the EDB-Fn targeting conjugate was significantly effective on EDB-Fn overexpressing cells, while the conjugate has minimal effect on nonexpressing or moderately expressing cells. In conclusion, it is plausible to state that the presence of conjugating moieties on compound 11 (integrin targeting) and compound 16 (EDB-Fn targeting) significantly affects their ability to adhere and deliver the chemotherapeutic agent selectively to biomarker overexpressing cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | HEK293 cell | CVCL_0045 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Prostate carcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
751.1 ± 27.2 nM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTS assay | ||||
| MOA of PDC |
The present study explores two different targeting peptides (TPs), RGDC (TP1), and CTVRTSAD (TP2), toward avβ1 integrin and EDB-Fn, respectively. TP1 or TP2 is covalently conjugated via disulfide bond to [C(WR)2K(WR)2]-(CBT) (6) to provide CBT conjugates (11, 16) containing an active targeting ability with increased aqueous solubility without decreasing cellular uptake. Targeting peptides may enable cCPP-CBT to target biomarker overexpressing tumor cells, and cCPP can assist in improving the physicochemical properties of CBT and delivery into the cancer cells. The conjugates in vitro stability was examined in various relevant conditions to mimic the cancerous versus normal physiological environment. Conjugates were evaluated in human plasma at two different glutathione concentrations (GSH, 1 mM and 10 mM) and two different pHs (6.5 and 7.4). The reported GSH level in noncancerous conditions is below 1 mM and approximately 10 mM at the cancer site. The in vitro antiproliferative assay using a biomarker overexpressing cell lines was compared to those without or with moderately expressing cancer cell lines. The study results demonstrate that targeting enhances the efficacy of CBT on cancer cell lines overexpressing extracellular biomarkers.
Click to Show/Hide
|
||||
| Description |
The conjugates therapeutic efficacy with targeting moieties was evaluated using prostate cancer cell lines with and without EDB-Fn overexpression (C4-2 and DU-145). Breast cancer cell lines with and without integrin v1 overexpression (MCF-7 and MDA-MB-231) were also used to evaluate antiproliferative activity. Noncancerous embryonic kidney epithelial cell line (HEK-293) was used as a control since this cell line did not express both biomarkers. All the compounds with and without targeting moieties showed comparable or lower antiproliferative activity than the CBT alone after 72 h incubation. The antiproliferative activity of compound 11 on MCF-7 (IC50 = 160.4 nM) was 1.6 times greater than on MDA-MB-231 (IC50 = 251.6 nM), showing a slight improvement in antiproliferative activity. A similar observation was found with compound 16 that showed 1.45 times higher antiproliferative activity on MCF-7 (IC50 = 162.4 nM) as compared to MDA-MB-231 cells (IC50 = 235.0 nM). Both MCF-7 and MDA-MB-231 are breast cancer cell lines. MDA-MB-231 is triple-negative breast cancer cells, and it showed higher IC50 values for CBT than MCF-7 cells. Later, the antiproliferative activity was compared between MCF-7 and the control cells (HEK-293); it was observed that compound 11 showed 3.33 times higher antiproliferative activity on MCF-7 cells (IC50 for 11 = 160.4 nM) compared to HEK-293 (IC50 for 11 = 534.5 nM), and compound 16 showed 4.6 times higher antiproliferative activity on MCF-7 (IC50 for 16 = 162.4 nM) compared to HEK-293 (IC50 for 16 = 483.3 nM). Thereafter, we wanted to assess the effectiveness of the EDB-Fn targeting moiety by comparing the antiproliferative activities on prostate cancer cell lines with and without EDB-Fn overexpression. The data showed a similar trend to that observed in breast cancer cells. For instance, compound 11 showed 3.4 times higher antiproliferative activity on C4-2 (IC50 for 11 = 181.4 nM) compared to DU-145 (IC50 for 11 = 611.2 nM). Similarly, compound 16 showed 3.7 times higher antiproliferative activity on C4-2 (IC50 for 16 = 202.2 nM) compared to DU-145 (IC50 for 16 = 751.1 nM). Compound 16 (possessing EDB-Fn targeting moiety) showed 1.1 times less antiproliferative activity compared to compound 6 (cCPP-CBT, without targeting moiety) on C4-2 cell lines, but compound 16 showed a much higher difference (3.6 times) in antiproliferative activity when compared with compound 6 on EDB-Fn moderately expressing DU-145 cell line. This data is further corroborated in EDB-Fn nonexpressing HEK-293. For instance, compound 16 showed 4.6 times less antiproliferative activity when compared with compound 6 on HEK-293 cell line. It can be inferred from the data (Table 2) that the EDB-Fn targeting conjugate was significantly effective on EDB-Fn overexpressing cells, while the conjugate has minimal effect on nonexpressing or moderately expressing cells. In conclusion, it is plausible to state that the presence of conjugating moieties on compound 11 (integrin targeting) and compound 16 (EDB-Fn targeting) significantly affects their ability to adhere and deliver the chemotherapeutic agent selectively to biomarker overexpressing cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | DU145 cell | CVCL_0105 | ||
[8Lys(Dox-O-glut)]-GnRH-III [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
0.1 ± 0.1 µM
|
|||
| Administration Time | 72 h | ||||
| MOA of PDC |
The efficacy of current chemotherapeutic treatments against most solid tumors is limited by their systemic toxicity, which is partly associated with the cytotoxic properties of agents such as docetaxel or doxorubicin. To avoid or minimize adverse effects from chemotherapeutic molecules, a promising targeted approach is through peptide-drug conjugates (PDCs) that link anticancer molecules to peptides designed to interact with receptors highly expressed on cancer cells, and which can mediate the molecules rapid internalization within those cells. One such receptor is sortilin (SORT1), also known as neurotensin receptor3, a membranebound receptor that belongs to the VPS10P family of receptors. TH19P01 peptide was recently designed to target and exploit SORT1s ligand internalization function. Studies have confirmed that both TH1902 (a docetaxel-TH19P01 conjugate) and TH1904 (a doxorubicin-TH19P01 conjugate) require a SORT1-dependent mechanism of action to exert anticancer activities. In recent preclinical studies performed in immunocompromised animal models, which are unable to produce mature T-cells, TH1902 was effective against several human SORT1-positive xenograft models including triple-negative breast cancer (TNBC), ovarian cancer, and endometrial cancer.
Click to Show/Hide
|
||||
| Description |
The main drug-related toxic side effect of anthracyclines is cardiotoxicity, leading to cardiomyopathy and heart failure. Comparative experiments to determine the cardiotoxicity of peptide-anthracycline conjugates with different linkages might be informative for the other conjugates with different types of drugs as well. For this purpose, human cardiomyocytes (HCM) and human umbilical vein endothelial cells (HUVEC) were used as models. The long-term (0-72 h) cytotoxic effect of sixteen GnRH-based conjugates containing Dox and Dau was determined by real-time impedimetric sensing using the xCELLigence SP system (ACEA Biosciences, San Diego, CA, USA) [75]. The results indicated that the ester-linked GnRH-Dox conjugates, including Zoptarelin Doxorubicin, showed significant toxicity at 100 nM and 1 uM, which was remarkably pronounced on the HCM cells. The cytotoxic effect was comparable to that of the free drug, especially at the highest concentration. In contrast, the conjugates with oxime-linked Dau showed no or only a minor toxicity on both the cell lines (Table 2). These data confirm that the linkage between the payload and homing peptide has a significant influence on early drug release and, consequently, an undesired toxic side effect. We may also conclude that the search for more suitable homing peptides might be more important than the application of cleavable bonds between the drug and the peptide to develop efficient DDSs.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.4 ± 0.2 µM
|
|||
| Administration Time | 72 h | ||||
| MOA of PDC |
The crucial steps during the synthesis of aminooxyacetylated peptides are the incorporation of aminooxyacetic acid (Aoa), including a protecting group, and the final cleavage and the working-up procedure of the Aoa-containing peptide derivatives. In most cases, Boc-protected Aoa is attached to a peptide chain in the last step of solid-phase peptide synthesis. It has been observed that over-acylation (additional Boc-Aoa-OH connected to the Aoa moiety) may occur as the main side reaction. Many approaches have been investigated to overcome this problem, including carbodiimide-mediated one-pot acylation without a base or the application of Boc-Aoa-OSu active ester as an acylating agent, as well as the use of a high excess (8 equiv) of Boc-Aoa-OH and coupling agents for a short acylation time (10 min). Nevertheless, the coupling of the diBoc-protected Aoa derivative has proved to be the best solution. However, the aminooxyacetyl moiety is very sensitive to molecules containing carbonyl groups, with the partial impact of the peptide sequence. Therefore, the free NH2-O-R group reacts often with these compounds during the working-up procedure after the final cleavage of Aoa-modified peptides from the resin. These carbonyl group-containing derivatives might come from the plastic tubes or residues of acetone used in a laboratory. This cannot be prevented even by using diBoc-protected Aoa or working in argon. We found a highly sensitive peptide to this side reaction; the synthesis of a somatostatin analog developed in Schallys laboratory elongated with Aoa (H-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2) was unsuccessful. After several trials to optimize the reaction conditions, we elaborated the following procedure: The semi-protected peptide H-D-Phe-Cys-Tyr-D-Trp-Lys(Dde)-Val-Cys-Thr-NH2 was cleaved from Rink Amide MBHA resin and reacted with Boc-Aoa-OPcp to incorporate Aoa into the N-terminus in a solution. After the efficient coupling reaction, the Dde-protecting group was removed with 2% hydrazine in DMF. Surprisingly, during the cleavage of Dde, a cyclic peptide also formed (Boc-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2). The Boc group was cleaved in 95% TFA solution in the presence of 10 equiv-free Aoa as a carbonyl capture reagent that could prevent the reaction of the peptide with any carbonyl derivative. The crude product was purified by RP-HPLC, and the solvent was evaporated, followed by direct ligation to daunomycin (Dau) in 0.2 M NaOAc solution at pH 5. This procedure proved to be very efficient to prepare oxime-linked Dau-peptide conjugates.
Click to Show/Hide
|
||||
| Description |
The main drug-related toxic side effect of anthracyclines is cardiotoxicity, leading to cardiomyopathy and heart failure. Comparative experiments to determine the cardiotoxicity of peptide-anthracycline conjugates with different linkages might be informative for the other conjugates with different types of drugs as well. For this purpose, human cardiomyocytes (HCM) and human umbilical vein endothelial cells (HUVEC) were used as models. The long-term (0-72 h) cytotoxic effect of sixteen GnRH-based conjugates containing Dox and Dau was determined by real-time impedimetric sensing using the xCELLigence SP system (ACEA Biosciences, San Diego, CA, USA) [75]. The results indicated that the ester-linked GnRH-Dox conjugates, including Zoptarelin Doxorubicin, showed significant toxicity at 100 nM and 1 uM, which was remarkably pronounced on the HCM cells. The cytotoxic effect was comparable to that of the free drug, especially at the highest concentration. In contrast, the conjugates with oxime-linked Dau showed no or only a minor toxicity on both the cell lines (Table 2). These data confirm that the linkage between the payload and homing peptide has a significant influence on early drug release and, consequently, an undesired toxic side effect. We may also conclude that the search for more suitable homing peptides might be more important than the application of cleavable bonds between the drug and the peptide to develop efficient DDSs.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
Peptide 18-4 doxorubicin conjugate 1 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [9] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
0.9 ± 0.07 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Here we report the design and synthesis of two new peptide-Dox conjugates (1 and 2) for the specific delivery of Dox to the breast cancer cells and the ability to overcome P-glycoprotein multidrug resistance pathway in both drug-sensitive and drug-resistant cancer cells. Peptide-Dox conjugates were evaluated for DOX release in human serum, intracellular delivery compared to free Dox in three cancerous cells (MCF-7, MDA-MB-435, and MDA-MB-435-MDR) and two noncancerous cell lines (HUVEC and MCF-10A), and cytotoxicity compared to free Dox. Results show that both the peptide-Dox conjugates (1 and 2) enter sensitive and resistant cell lines with minimal uptake in normal cells compared to free Dox. Cellular uptake is most likely mediated by a cell specific receptor, as the amount of internalized conjugates significantly decreased in the presence of excess free peptide. Importantly, conjugate 1 is equally cytotoxic as Dox in drug sensitive breast cancer cells and 4 times more potent than free Dox in Dox resistant cell line. Overall, the peptide-Dox ester conjugate 1 showed better breast targeting efficacy than the amide conjugate 2, most likely due to the slow release of Dox from the stable amide linkage.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity experiment was done by incubating the cells with different treatments for 48 h. The results show that conjugate 1 is quite similar to free Dox for toxicity to MCF-7 and MDA-MB-435 cancer cells. In contrast, conjugate 2 was ?20 times less cytotoxic to breast cancerous cells compared to free Dox. This could be attributed to the stability of the amide conjugate 2 inside the cells. Furthermore, the cytotoxicity of conjugate 1 in doxorubicin-resistant cell model MDA-MB-435-MDR (IC50 = 5.4 μM) is 4 times more than free Dox (IC50 = 22 μM). In normal cells (HUVEC and MCF-10A) the two conjugates were 3540 times less toxic compared to breast cancer cells, whereas free Dox was equally cytotoxic (equal IC50) to breast cancer cells and noncancerous cells. Overall these results provide clear evidence that of the two conjugates (conjugate 1 and 2), conjugate 1 has optimal characteristics for specific Dox targeting to breast cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [9] | ||||
| Indication | Amelanotic melanoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
1.5 ± 0.09 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Here we report the design and synthesis of two new peptide-Dox conjugates (1 and 2) for the specific delivery of Dox to the breast cancer cells and the ability to overcome P-glycoprotein multidrug resistance pathway in both drug-sensitive and drug-resistant cancer cells. Peptide-Dox conjugates were evaluated for DOX release in human serum, intracellular delivery compared to free Dox in three cancerous cells (MCF-7, MDA-MB-435, and MDA-MB-435-MDR) and two noncancerous cell lines (HUVEC and MCF-10A), and cytotoxicity compared to free Dox. Results show that both the peptide-Dox conjugates (1 and 2) enter sensitive and resistant cell lines with minimal uptake in normal cells compared to free Dox. Cellular uptake is most likely mediated by a cell specific receptor, as the amount of internalized conjugates significantly decreased in the presence of excess free peptide. Importantly, conjugate 1 is equally cytotoxic as Dox in drug sensitive breast cancer cells and 4 times more potent than free Dox in Dox resistant cell line. Overall, the peptide-Dox ester conjugate 1 showed better breast targeting efficacy than the amide conjugate 2, most likely due to the slow release of Dox from the stable amide linkage.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity experiment was done by incubating the cells with different treatments for 48 h. The results show that conjugate 1 is quite similar to free Dox for toxicity to MCF-7 and MDA-MB-435 cancer cells. In contrast, conjugate 2 was ?20 times less cytotoxic to breast cancerous cells compared to free Dox. This could be attributed to the stability of the amide conjugate 2 inside the cells. Furthermore, the cytotoxicity of conjugate 1 in doxorubicin-resistant cell model MDA-MB-435-MDR (IC50 = 5.4 μM) is 4 times more than free Dox (IC50 = 22 μM). In normal cells (HUVEC and MCF-10A) the two conjugates were 3540 times less toxic compared to breast cancer cells, whereas free Dox was equally cytotoxic (equal IC50) to breast cancer cells and noncancerous cells. Overall these results provide clear evidence that of the two conjugates (conjugate 1 and 2), conjugate 1 has optimal characteristics for specific Dox targeting to breast cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Amelanotic melanoma | MDA-MB-435 cell | CVCL_0417 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [9] | ||||
| Indication | Amelanotic melanoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
5.4 ± 0.62 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Here we report the design and synthesis of two new peptide-Dox conjugates (1 and 2) for the specific delivery of Dox to the breast cancer cells and the ability to overcome P-glycoprotein multidrug resistance pathway in both drug-sensitive and drug-resistant cancer cells. Peptide-Dox conjugates were evaluated for DOX release in human serum, intracellular delivery compared to free Dox in three cancerous cells (MCF-7, MDA-MB-435, and MDA-MB-435-MDR) and two noncancerous cell lines (HUVEC and MCF-10A), and cytotoxicity compared to free Dox. Results show that both the peptide-Dox conjugates (1 and 2) enter sensitive and resistant cell lines with minimal uptake in normal cells compared to free Dox. Cellular uptake is most likely mediated by a cell specific receptor, as the amount of internalized conjugates significantly decreased in the presence of excess free peptide. Importantly, conjugate 1 is equally cytotoxic as Dox in drug sensitive breast cancer cells and 4 times more potent than free Dox in Dox resistant cell line. Overall, the peptide-Dox ester conjugate 1 showed better breast targeting efficacy than the amide conjugate 2, most likely due to the slow release of Dox from the stable amide linkage.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity experiment was done by incubating the cells with different treatments for 48 h. The results show that conjugate 1 is quite similar to free Dox for toxicity to MCF-7 and MDA-MB-435 cancer cells. In contrast, conjugate 2 was ?20 times less cytotoxic to breast cancerous cells compared to free Dox. This could be attributed to the stability of the amide conjugate 2 inside the cells. Furthermore, the cytotoxicity of conjugate 1 in doxorubicin-resistant cell model MDA-MB-435-MDR (IC50 = 5.4 μM) is 4 times more than free Dox (IC50 = 22 μM). In normal cells (HUVEC and MCF-10A) the two conjugates were 3540 times less toxic compared to breast cancer cells, whereas free Dox was equally cytotoxic (equal IC50) to breast cancer cells and noncancerous cells. Overall these results provide clear evidence that of the two conjugates (conjugate 1 and 2), conjugate 1 has optimal characteristics for specific Dox targeting to breast cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Amelanotic melanoma | MDA-MB-435 cell | CVCL_0417 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [9] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
35.1 ± 2.2 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Here we report the design and synthesis of two new peptide-Dox conjugates (1 and 2) for the specific delivery of Dox to the breast cancer cells and the ability to overcome P-glycoprotein multidrug resistance pathway in both drug-sensitive and drug-resistant cancer cells. Peptide-Dox conjugates were evaluated for DOX release in human serum, intracellular delivery compared to free Dox in three cancerous cells (MCF-7, MDA-MB-435, and MDA-MB-435-MDR) and two noncancerous cell lines (HUVEC and MCF-10A), and cytotoxicity compared to free Dox. Results show that both the peptide-Dox conjugates (1 and 2) enter sensitive and resistant cell lines with minimal uptake in normal cells compared to free Dox. Cellular uptake is most likely mediated by a cell specific receptor, as the amount of internalized conjugates significantly decreased in the presence of excess free peptide. Importantly, conjugate 1 is equally cytotoxic as Dox in drug sensitive breast cancer cells and 4 times more potent than free Dox in Dox resistant cell line. Overall, the peptide-Dox ester conjugate 1 showed better breast targeting efficacy than the amide conjugate 2, most likely due to the slow release of Dox from the stable amide linkage.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity experiment was done by incubating the cells with different treatments for 48 h. The results show that conjugate 1 is quite similar to free Dox for toxicity to MCF-7 and MDA-MB-435 cancer cells. In contrast, conjugate 2 was ?20 times less cytotoxic to breast cancerous cells compared to free Dox. This could be attributed to the stability of the amide conjugate 2 inside the cells. Furthermore, the cytotoxicity of conjugate 1 in doxorubicin-resistant cell model MDA-MB-435-MDR (IC50 = 5.4 μM) is 4 times more than free Dox (IC50 = 22 μM). In normal cells (HUVEC and MCF-10A) the two conjugates were 3540 times less toxic compared to breast cancer cells, whereas free Dox was equally cytotoxic (equal IC50) to breast cancer cells and noncancerous cells. Overall these results provide clear evidence that of the two conjugates (conjugate 1 and 2), conjugate 1 has optimal characteristics for specific Dox targeting to breast cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [9] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
42.3 ± 2.4 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Here we report the design and synthesis of two new peptide-Dox conjugates (1 and 2) for the specific delivery of Dox to the breast cancer cells and the ability to overcome P-glycoprotein multidrug resistance pathway in both drug-sensitive and drug-resistant cancer cells. Peptide-Dox conjugates were evaluated for DOX release in human serum, intracellular delivery compared to free Dox in three cancerous cells (MCF-7, MDA-MB-435, and MDA-MB-435-MDR) and two noncancerous cell lines (HUVEC and MCF-10A), and cytotoxicity compared to free Dox. Results show that both the peptide-Dox conjugates (1 and 2) enter sensitive and resistant cell lines with minimal uptake in normal cells compared to free Dox. Cellular uptake is most likely mediated by a cell specific receptor, as the amount of internalized conjugates significantly decreased in the presence of excess free peptide. Importantly, conjugate 1 is equally cytotoxic as Dox in drug sensitive breast cancer cells and 4 times more potent than free Dox in Dox resistant cell line. Overall, the peptide-Dox ester conjugate 1 showed better breast targeting efficacy than the amide conjugate 2, most likely due to the slow release of Dox from the stable amide linkage.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity experiment was done by incubating the cells with different treatments for 48 h. The results show that conjugate 1 is quite similar to free Dox for toxicity to MCF-7 and MDA-MB-435 cancer cells. In contrast, conjugate 2 was ?20 times less cytotoxic to breast cancerous cells compared to free Dox. This could be attributed to the stability of the amide conjugate 2 inside the cells. Furthermore, the cytotoxicity of conjugate 1 in doxorubicin-resistant cell model MDA-MB-435-MDR (IC50 = 5.4 μM) is 4 times more than free Dox (IC50 = 22 μM). In normal cells (HUVEC and MCF-10A) the two conjugates were 3540 times less toxic compared to breast cancer cells, whereas free Dox was equally cytotoxic (equal IC50) to breast cancer cells and noncancerous cells. Overall these results provide clear evidence that of the two conjugates (conjugate 1 and 2), conjugate 1 has optimal characteristics for specific Dox targeting to breast cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Human umbilical vein endothelial cell | Homo sapiens | ||
R-C12-2 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
3.3 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, we would investigate the co-modification of fatty acids and monosaccharides in anticancer peptides, as we expect to optimize the selectivity and cytotoxicity of the peptide at the same time. Based on our previous study, the lipopeptide R-C12 was selected as a template to perform glucose derivative modification at different positions as we supposed that the glucose derivative modification position in R-C12 might be a crucial factor in its selectivity and activity optimization. The glucose derivative was covalently coupled to different sites in R-C12 through its -amino group of a Lys residue, which was obtained by replacing the Arg residue. R-C12 contains seven Arg residues, accordingly, seven glycolipid peptides were designed and synthesized, which were named R-C12-1, R-C12-2, R-C12-3, R-C12-4, R-C12-5, R-C12-6, and R-C12-7. Various physiochemical properties including the hydrodynamic size, ζ-potential, secondary structure, and hydrophobicity were determined to explain the different cytotoxicity and selectivity of these glycolipid peptides. On the other hand, the strength of the binding between glycolipid peptides and the cancer cells mediated by GLUT1 has also been explored. The most optimized glycolipid peptide R-C12-4 demonstrated significant cytotoxicity and antimetastasis in vitro and in vivo, which indicated that it may be a good lead for anticancer drug development.
Click to Show/Hide
|
||||
| Description |
Murine melanoma cells (B16-F10) were chosen as the object for evaluating the cytotoxicity of the seven glycolipid peptides and R-C12 by the CCK-8 assay. Based on our previous study, the HEK-293T cell line was also chosen as the noncancer cell line to evaluate the selectivity of glycolipid peptides. IC50 represents the concentration of glycolipid peptides and R-C12 that induce 50% cell death. As shown in Figure 1A, modification of the glucose derivative at different sites of R-C12 results in different cytotoxicity profiles. According to the IC50 values, for B16-F10 cells, the cytotoxic activity of peptides was arranged as follows R-C12-4 > R-C12-1 ≈ R-C12-2 ≈ R-C12-3 ≈ R-C12 > R-C12-5 > R-C12-6 > R-C12-7, in which R-C12-4 demonstrated the highest cytotoxicity on B16-F10 cells (IC50 = 1.9 μM)
Click to Show/Hide
|
||||
| In Vitro Model | Mouse melanoma | B16-F10 cell | CVCL_0159 | ||
R-C12-1 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
3.5 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, we would investigate the co-modification of fatty acids and monosaccharides in anticancer peptides, as we expect to optimize the selectivity and cytotoxicity of the peptide at the same time. Based on our previous study, the lipopeptide R-C12 was selected as a template to perform glucose derivative modification at different positions as we supposed that the glucose derivative modification position in R-C12 might be a crucial factor in its selectivity and activity optimization. The glucose derivative was covalently coupled to different sites in R-C12 through its -amino group of a Lys residue, which was obtained by replacing the Arg residue. R-C12 contains seven Arg residues, accordingly, seven glycolipid peptides were designed and synthesized, which were named R-C12-1, R-C12-2, R-C12-3, R-C12-4, R-C12-5, R-C12-6, and R-C12-7. Various physiochemical properties including the hydrodynamic size, ζ-potential, secondary structure, and hydrophobicity were determined to explain the different cytotoxicity and selectivity of these glycolipid peptides. On the other hand, the strength of the binding between glycolipid peptides and the cancer cells mediated by GLUT1 has also been explored. The most optimized glycolipid peptide R-C12-4 demonstrated significant cytotoxicity and antimetastasis in vitro and in vivo, which indicated that it may be a good lead for anticancer drug development.
Click to Show/Hide
|
||||
| Description |
Murine melanoma cells (B16-F10) were chosen as the object for evaluating the cytotoxicity of the seven glycolipid peptides and R-C12 by the CCK-8 assay. Based on our previous study, the HEK-293T cell line was also chosen as the noncancer cell line to evaluate the selectivity of glycolipid peptides. IC50 represents the concentration of glycolipid peptides and R-C12 that induce 50% cell death. As shown in Figure 1A, modification of the glucose derivative at different sites of R-C12 results in different cytotoxicity profiles. According to the IC50 values, for B16-F10 cells, the cytotoxic activity of peptides was arranged as follows R-C12-4 > R-C12-1 ≈ R-C12-2 ≈ R-C12-3 ≈ R-C12 > R-C12-5 > R-C12-6 > R-C12-7, in which R-C12-4 demonstrated the highest cytotoxicity on B16-F10 cells (IC50 = 1.9 μM)
Click to Show/Hide
|
||||
| In Vitro Model | Mouse melanoma | B16-F10 cell | CVCL_0159 | ||
R-C12-3 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
3.8 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, we would investigate the co-modification of fatty acids and monosaccharides in anticancer peptides, as we expect to optimize the selectivity and cytotoxicity of the peptide at the same time. Based on our previous study, the lipopeptide R-C12 was selected as a template to perform glucose derivative modification at different positions as we supposed that the glucose derivative modification position in R-C12 might be a crucial factor in its selectivity and activity optimization. The glucose derivative was covalently coupled to different sites in R-C12 through its -amino group of a Lys residue, which was obtained by replacing the Arg residue. R-C12 contains seven Arg residues, accordingly, seven glycolipid peptides were designed and synthesized, which were named R-C12-1, R-C12-2, R-C12-3, R-C12-4, R-C12-5, R-C12-6, and R-C12-7. Various physiochemical properties including the hydrodynamic size, ζ-potential, secondary structure, and hydrophobicity were determined to explain the different cytotoxicity and selectivity of these glycolipid peptides. On the other hand, the strength of the binding between glycolipid peptides and the cancer cells mediated by GLUT1 has also been explored. The most optimized glycolipid peptide R-C12-4 demonstrated significant cytotoxicity and antimetastasis in vitro and in vivo, which indicated that it may be a good lead for anticancer drug development.
Click to Show/Hide
|
||||
| Description |
Murine melanoma cells (B16-F10) were chosen as the object for evaluating the cytotoxicity of the seven glycolipid peptides and R-C12 by the CCK-8 assay. Based on our previous study, the HEK-293T cell line was also chosen as the noncancer cell line to evaluate the selectivity of glycolipid peptides. IC50 represents the concentration of glycolipid peptides and R-C12 that induce 50% cell death. As shown in Figure 1A, modification of the glucose derivative at different sites of R-C12 results in different cytotoxicity profiles. According to the IC50 values, for B16-F10 cells, the cytotoxic activity of peptides was arranged as follows R-C12-4 > R-C12-1 ≈ R-C12-2 ≈ R-C12-3 ≈ R-C12 > R-C12-5 > R-C12-6 > R-C12-7, in which R-C12-4 demonstrated the highest cytotoxicity on B16-F10 cells (IC50 = 1.9 μM)
Click to Show/Hide
|
||||
| In Vitro Model | Mouse melanoma | B16-F10 cell | CVCL_0159 | ||
R-C12-5 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
4.1 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, we would investigate the co-modification of fatty acids and monosaccharides in anticancer peptides, as we expect to optimize the selectivity and cytotoxicity of the peptide at the same time. Based on our previous study, the lipopeptide R-C12 was selected as a template to perform glucose derivative modification at different positions as we supposed that the glucose derivative modification position in R-C12 might be a crucial factor in its selectivity and activity optimization. The glucose derivative was covalently coupled to different sites in R-C12 through its -amino group of a Lys residue, which was obtained by replacing the Arg residue. R-C12 contains seven Arg residues, accordingly, seven glycolipid peptides were designed and synthesized, which were named R-C12-1, R-C12-2, R-C12-3, R-C12-4, R-C12-5, R-C12-6, and R-C12-7. Various physiochemical properties including the hydrodynamic size, ζ-potential, secondary structure, and hydrophobicity were determined to explain the different cytotoxicity and selectivity of these glycolipid peptides. On the other hand, the strength of the binding between glycolipid peptides and the cancer cells mediated by GLUT1 has also been explored. The most optimized glycolipid peptide R-C12-4 demonstrated significant cytotoxicity and antimetastasis in vitro and in vivo, which indicated that it may be a good lead for anticancer drug development.
Click to Show/Hide
|
||||
| Description |
Murine melanoma cells (B16-F10) were chosen as the object for evaluating the cytotoxicity of the seven glycolipid peptides and R-C12 by the CCK-8 assay. Based on our previous study, the HEK-293T cell line was also chosen as the noncancer cell line to evaluate the selectivity of glycolipid peptides. IC50 represents the concentration of glycolipid peptides and R-C12 that induce 50% cell death. As shown in Figure 1A, modification of the glucose derivative at different sites of R-C12 results in different cytotoxicity profiles. According to the IC50 values, for B16-F10 cells, the cytotoxic activity of peptides was arranged as follows R-C12-4 > R-C12-1 ≈ R-C12-2 ≈ R-C12-3 ≈ R-C12 > R-C12-5 > R-C12-6 > R-C12-7, in which R-C12-4 demonstrated the highest cytotoxicity on B16-F10 cells (IC50 = 1.9 μM)
Click to Show/Hide
|
||||
| In Vitro Model | Mouse melanoma | B16-F10 cell | CVCL_0159 | ||
R-C12-6 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
5.3 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, we would investigate the co-modification of fatty acids and monosaccharides in anticancer peptides, as we expect to optimize the selectivity and cytotoxicity of the peptide at the same time. Based on our previous study, the lipopeptide R-C12 was selected as a template to perform glucose derivative modification at different positions as we supposed that the glucose derivative modification position in R-C12 might be a crucial factor in its selectivity and activity optimization. The glucose derivative was covalently coupled to different sites in R-C12 through its -amino group of a Lys residue, which was obtained by replacing the Arg residue. R-C12 contains seven Arg residues, accordingly, seven glycolipid peptides were designed and synthesized, which were named R-C12-1, R-C12-2, R-C12-3, R-C12-4, R-C12-5, R-C12-6, and R-C12-7. Various physiochemical properties including the hydrodynamic size, ζ-potential, secondary structure, and hydrophobicity were determined to explain the different cytotoxicity and selectivity of these glycolipid peptides. On the other hand, the strength of the binding between glycolipid peptides and the cancer cells mediated by GLUT1 has also been explored. The most optimized glycolipid peptide R-C12-4 demonstrated significant cytotoxicity and antimetastasis in vitro and in vivo, which indicated that it may be a good lead for anticancer drug development.
Click to Show/Hide
|
||||
| Description |
Murine melanoma cells (B16-F10) were chosen as the object for evaluating the cytotoxicity of the seven glycolipid peptides and R-C12 by the CCK-8 assay. Based on our previous study, the HEK-293T cell line was also chosen as the noncancer cell line to evaluate the selectivity of glycolipid peptides. IC50 represents the concentration of glycolipid peptides and R-C12 that induce 50% cell death. As shown in Figure 1A, modification of the glucose derivative at different sites of R-C12 results in different cytotoxicity profiles. According to the IC50 values, for B16-F10 cells, the cytotoxic activity of peptides was arranged as follows R-C12-4 > R-C12-1 ≈ R-C12-2 ≈ R-C12-3 ≈ R-C12 > R-C12-5 > R-C12-6 > R-C12-7, in which R-C12-4 demonstrated the highest cytotoxicity on B16-F10 cells (IC50 = 1.9 μM)
Click to Show/Hide
|
||||
| In Vitro Model | Mouse melanoma | B16-F10 cell | CVCL_0159 | ||
R-C12-7 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
7.2 μM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, we would investigate the co-modification of fatty acids and monosaccharides in anticancer peptides, as we expect to optimize the selectivity and cytotoxicity of the peptide at the same time. Based on our previous study, the lipopeptide R-C12 was selected as a template to perform glucose derivative modification at different positions as we supposed that the glucose derivative modification position in R-C12 might be a crucial factor in its selectivity and activity optimization. The glucose derivative was covalently coupled to different sites in R-C12 through its -amino group of a Lys residue, which was obtained by replacing the Arg residue. R-C12 contains seven Arg residues, accordingly, seven glycolipid peptides were designed and synthesized, which were named R-C12-1, R-C12-2, R-C12-3, R-C12-4, R-C12-5, R-C12-6, and R-C12-7. Various physiochemical properties including the hydrodynamic size, ζ-potential, secondary structure, and hydrophobicity were determined to explain the different cytotoxicity and selectivity of these glycolipid peptides. On the other hand, the strength of the binding between glycolipid peptides and the cancer cells mediated by GLUT1 has also been explored. The most optimized glycolipid peptide R-C12-4 demonstrated significant cytotoxicity and antimetastasis in vitro and in vivo, which indicated that it may be a good lead for anticancer drug development.
Click to Show/Hide
|
||||
| Description |
Murine melanoma cells (B16-F10) were chosen as the object for evaluating the cytotoxicity of the seven glycolipid peptides and R-C12 by the CCK-8 assay. Based on our previous study, the HEK-293T cell line was also chosen as the noncancer cell line to evaluate the selectivity of glycolipid peptides. IC50 represents the concentration of glycolipid peptides and R-C12 that induce 50% cell death. As shown in Figure 1A, modification of the glucose derivative at different sites of R-C12 results in different cytotoxicity profiles. According to the IC50 values, for B16-F10 cells, the cytotoxic activity of peptides was arranged as follows R-C12-4 > R-C12-1 ≈ R-C12-2 ≈ R-C12-3 ≈ R-C12 > R-C12-5 > R-C12-6 > R-C12-7, in which R-C12-4 demonstrated the highest cytotoxicity on B16-F10 cells (IC50 = 1.9 μM)
Click to Show/Hide
|
||||
| In Vitro Model | Mouse melanoma | B16-F10 cell | CVCL_0159 | ||
GnRH-III-[2His-3Trp,8Lys(glutaryl-Val-Cit-PABC-diamine-PTX)] conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [10] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
8.61 ± 1.08 µM
|
|||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate cancer | Human prostate cancer cells | Homo sapiens | ||
| Experiment 2 Reporting the Activity Data of This PDC | [10] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
9.70 ± 1.07 µM
|
|||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Normal human pituitary cell | Homo sapiens | ||
| Experiment 3 Reporting the Activity Data of This PDC | [10] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
67.88 ± 25.36 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | GraphPad prism assay | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [10] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 100 µM | |||
| Administration Time | 72 h | ||||
| Evaluation Method | GraphPad prism assay | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | PANC-1 cell | CVCL_0480 | ||
Peptide 18-4 doxorubicin conjugate 2 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [9] | ||||
| Indication | Amelanotic melanoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
18.6 ± 2.5 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Here we report the design and synthesis of two new peptide-Dox conjugates (1 and 2) for the specific delivery of Dox to the breast cancer cells and the ability to overcome P-glycoprotein multidrug resistance pathway in both drug-sensitive and drug-resistant cancer cells. Peptide-Dox conjugates were evaluated for DOX release in human serum, intracellular delivery compared to free Dox in three cancerous cells (MCF-7, MDA-MB-435, and MDA-MB-435-MDR) and two noncancerous cell lines (HUVEC and MCF-10A), and cytotoxicity compared to free Dox. Results show that both the peptide-Dox conjugates (1 and 2) enter sensitive and resistant cell lines with minimal uptake in normal cells compared to free Dox. Cellular uptake is most likely mediated by a cell specific receptor, as the amount of internalized conjugates significantly decreased in the presence of excess free peptide. Importantly, conjugate 1 is equally cytotoxic as Dox in drug sensitive breast cancer cells and 4 times more potent than free Dox in Dox resistant cell line. Overall, the peptide-Dox ester conjugate 1 showed better breast targeting efficacy than the amide conjugate 2, most likely due to the slow release of Dox from the stable amide linkage.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity experiment was done by incubating the cells with different treatments for 48 h. The results show that conjugate 1 is quite similar to free Dox for toxicity to MCF-7 and MDA-MB-435 cancer cells. In contrast, conjugate 2 was ?20 times less cytotoxic to breast cancerous cells compared to free Dox. This could be attributed to the stability of the amide conjugate 2 inside the cells. Furthermore, the cytotoxicity of conjugate 1 in doxorubicin-resistant cell model MDA-MB-435-MDR (IC50 = 5.4 μM) is 4 times more than free Dox (IC50 = 22 μM). In normal cells (HUVEC and MCF-10A) the two conjugates were 3540 times less toxic compared to breast cancer cells, whereas free Dox was equally cytotoxic (equal IC50) to breast cancer cells and noncancerous cells. Overall these results provide clear evidence that of the two conjugates (conjugate 1 and 2), conjugate 1 has optimal characteristics for specific Dox targeting to breast cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Amelanotic melanoma | MDA-MB-435 cell | CVCL_0417 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [9] | ||||
| Indication | Amelanotic melanoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
19.7 ± 3.1 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Here we report the design and synthesis of two new peptide-Dox conjugates (1 and 2) for the specific delivery of Dox to the breast cancer cells and the ability to overcome P-glycoprotein multidrug resistance pathway in both drug-sensitive and drug-resistant cancer cells. Peptide-Dox conjugates were evaluated for DOX release in human serum, intracellular delivery compared to free Dox in three cancerous cells (MCF-7, MDA-MB-435, and MDA-MB-435-MDR) and two noncancerous cell lines (HUVEC and MCF-10A), and cytotoxicity compared to free Dox. Results show that both the peptide-Dox conjugates (1 and 2) enter sensitive and resistant cell lines with minimal uptake in normal cells compared to free Dox. Cellular uptake is most likely mediated by a cell specific receptor, as the amount of internalized conjugates significantly decreased in the presence of excess free peptide. Importantly, conjugate 1 is equally cytotoxic as Dox in drug sensitive breast cancer cells and 4 times more potent than free Dox in Dox resistant cell line. Overall, the peptide-Dox ester conjugate 1 showed better breast targeting efficacy than the amide conjugate 2, most likely due to the slow release of Dox from the stable amide linkage.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity experiment was done by incubating the cells with different treatments for 48 h. The results show that conjugate 1 is quite similar to free Dox for toxicity to MCF-7 and MDA-MB-435 cancer cells. In contrast, conjugate 2 was ?20 times less cytotoxic to breast cancerous cells compared to free Dox. This could be attributed to the stability of the amide conjugate 2 inside the cells. Furthermore, the cytotoxicity of conjugate 1 in doxorubicin-resistant cell model MDA-MB-435-MDR (IC50 = 5.4 μM) is 4 times more than free Dox (IC50 = 22 μM). In normal cells (HUVEC and MCF-10A) the two conjugates were 3540 times less toxic compared to breast cancer cells, whereas free Dox was equally cytotoxic (equal IC50) to breast cancer cells and noncancerous cells. Overall these results provide clear evidence that of the two conjugates (conjugate 1 and 2), conjugate 1 has optimal characteristics for specific Dox targeting to breast cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Amelanotic melanoma | MDA-MB-435 cell | CVCL_0417 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [9] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
40.5 ± 4.3 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Here we report the design and synthesis of two new peptide-Dox conjugates (1 and 2) for the specific delivery of Dox to the breast cancer cells and the ability to overcome P-glycoprotein multidrug resistance pathway in both drug-sensitive and drug-resistant cancer cells. Peptide-Dox conjugates were evaluated for DOX release in human serum, intracellular delivery compared to free Dox in three cancerous cells (MCF-7, MDA-MB-435, and MDA-MB-435-MDR) and two noncancerous cell lines (HUVEC and MCF-10A), and cytotoxicity compared to free Dox. Results show that both the peptide-Dox conjugates (1 and 2) enter sensitive and resistant cell lines with minimal uptake in normal cells compared to free Dox. Cellular uptake is most likely mediated by a cell specific receptor, as the amount of internalized conjugates significantly decreased in the presence of excess free peptide. Importantly, conjugate 1 is equally cytotoxic as Dox in drug sensitive breast cancer cells and 4 times more potent than free Dox in Dox resistant cell line. Overall, the peptide-Dox ester conjugate 1 showed better breast targeting efficacy than the amide conjugate 2, most likely due to the slow release of Dox from the stable amide linkage.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity experiment was done by incubating the cells with different treatments for 48 h. The results show that conjugate 1 is quite similar to free Dox for toxicity to MCF-7 and MDA-MB-435 cancer cells. In contrast, conjugate 2 was ?20 times less cytotoxic to breast cancerous cells compared to free Dox. This could be attributed to the stability of the amide conjugate 2 inside the cells. Furthermore, the cytotoxicity of conjugate 1 in doxorubicin-resistant cell model MDA-MB-435-MDR (IC50 = 5.4 μM) is 4 times more than free Dox (IC50 = 22 μM). In normal cells (HUVEC and MCF-10A) the two conjugates were 3540 times less toxic compared to breast cancer cells, whereas free Dox was equally cytotoxic (equal IC50) to breast cancer cells and noncancerous cells. Overall these results provide clear evidence that of the two conjugates (conjugate 1 and 2), conjugate 1 has optimal characteristics for specific Dox targeting to breast cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [9] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
50.9 ± 3.2 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Here we report the design and synthesis of two new peptide-Dox conjugates (1 and 2) for the specific delivery of Dox to the breast cancer cells and the ability to overcome P-glycoprotein multidrug resistance pathway in both drug-sensitive and drug-resistant cancer cells. Peptide-Dox conjugates were evaluated for DOX release in human serum, intracellular delivery compared to free Dox in three cancerous cells (MCF-7, MDA-MB-435, and MDA-MB-435-MDR) and two noncancerous cell lines (HUVEC and MCF-10A), and cytotoxicity compared to free Dox. Results show that both the peptide-Dox conjugates (1 and 2) enter sensitive and resistant cell lines with minimal uptake in normal cells compared to free Dox. Cellular uptake is most likely mediated by a cell specific receptor, as the amount of internalized conjugates significantly decreased in the presence of excess free peptide. Importantly, conjugate 1 is equally cytotoxic as Dox in drug sensitive breast cancer cells and 4 times more potent than free Dox in Dox resistant cell line. Overall, the peptide-Dox ester conjugate 1 showed better breast targeting efficacy than the amide conjugate 2, most likely due to the slow release of Dox from the stable amide linkage.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity experiment was done by incubating the cells with different treatments for 48 h. The results show that conjugate 1 is quite similar to free Dox for toxicity to MCF-7 and MDA-MB-435 cancer cells. In contrast, conjugate 2 was ?20 times less cytotoxic to breast cancerous cells compared to free Dox. This could be attributed to the stability of the amide conjugate 2 inside the cells. Furthermore, the cytotoxicity of conjugate 1 in doxorubicin-resistant cell model MDA-MB-435-MDR (IC50 = 5.4 μM) is 4 times more than free Dox (IC50 = 22 μM). In normal cells (HUVEC and MCF-10A) the two conjugates were 3540 times less toxic compared to breast cancer cells, whereas free Dox was equally cytotoxic (equal IC50) to breast cancer cells and noncancerous cells. Overall these results provide clear evidence that of the two conjugates (conjugate 1 and 2), conjugate 1 has optimal characteristics for specific Dox targeting to breast cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Human umbilical vein endothelial cell | Homo sapiens | ||
| Experiment 5 Reporting the Activity Data of This PDC | [9] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
191 ± 2.8 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Here we report the design and synthesis of two new peptide-Dox conjugates (1 and 2) for the specific delivery of Dox to the breast cancer cells and the ability to overcome P-glycoprotein multidrug resistance pathway in both drug-sensitive and drug-resistant cancer cells. Peptide-Dox conjugates were evaluated for DOX release in human serum, intracellular delivery compared to free Dox in three cancerous cells (MCF-7, MDA-MB-435, and MDA-MB-435-MDR) and two noncancerous cell lines (HUVEC and MCF-10A), and cytotoxicity compared to free Dox. Results show that both the peptide-Dox conjugates (1 and 2) enter sensitive and resistant cell lines with minimal uptake in normal cells compared to free Dox. Cellular uptake is most likely mediated by a cell specific receptor, as the amount of internalized conjugates significantly decreased in the presence of excess free peptide. Importantly, conjugate 1 is equally cytotoxic as Dox in drug sensitive breast cancer cells and 4 times more potent than free Dox in Dox resistant cell line. Overall, the peptide-Dox ester conjugate 1 showed better breast targeting efficacy than the amide conjugate 2, most likely due to the slow release of Dox from the stable amide linkage.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity experiment was done by incubating the cells with different treatments for 48 h. The results show that conjugate 1 is quite similar to free Dox for toxicity to MCF-7 and MDA-MB-435 cancer cells. In contrast, conjugate 2 was ?20 times less cytotoxic to breast cancerous cells compared to free Dox. This could be attributed to the stability of the amide conjugate 2 inside the cells. Furthermore, the cytotoxicity of conjugate 1 in doxorubicin-resistant cell model MDA-MB-435-MDR (IC50 = 5.4 μM) is 4 times more than free Dox (IC50 = 22 μM). In normal cells (HUVEC and MCF-10A) the two conjugates were 3540 times less toxic compared to breast cancer cells, whereas free Dox was equally cytotoxic (equal IC50) to breast cancer cells and noncancerous cells. Overall these results provide clear evidence that of the two conjugates (conjugate 1 and 2), conjugate 1 has optimal characteristics for specific Dox targeting to breast cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
GnRH-III-[2ΔHis-3D-Tic, 8Lys(glutaryl-diamine-PTX)] conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [10] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
22.21 ± 0.96 µM
|
|||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate cancer | Human prostate cancer cells | Homo sapiens | ||
| Experiment 2 Reporting the Activity Data of This PDC | [10] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
23.43 ± 1.67 µM
|
|||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Normal human pituitary cell | Homo sapiens | ||
| Experiment 3 Reporting the Activity Data of This PDC | [10] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 100 µM | |||
| Administration Time | 72 h | ||||
| Evaluation Method | GraphPad prism assay | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [10] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 100 µM | |||
| Administration Time | 72 h | ||||
| Evaluation Method | GraphPad prism assay | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | PANC-1 cell | CVCL_0480 | ||
GnRH-III-[2ΔHis-3D-Tic,8Lys(glutaryl-Val-Cit-PABC-diamine-PTX)] conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [10] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
36.29 ± 3.17 µM
|
|||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Normal human pituitary cell | Homo sapiens | ||
| Experiment 2 Reporting the Activity Data of This PDC | [10] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
42.67 ± 7.04 µM
|
|||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate cancer | Human prostate cancer cells | Homo sapiens | ||
| Experiment 3 Reporting the Activity Data of This PDC | [10] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
48.14 ± 0.47 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | GraphPad prism assay | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [10] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 100 µM | |||
| Administration Time | 72 h | ||||
| Evaluation Method | GraphPad prism assay | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | PANC-1 cell | CVCL_0480 | ||
GnRH-III-[2His-3Trp,8Lys(glutaryl-diamine-PTX)] conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [10] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
41.52 ± 9.83 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | GraphPad prism assay | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [10] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 100 µM | |||
| Administration Time | 72 h | ||||
| Evaluation Method | GraphPad prism assay | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | PANC-1 cell | CVCL_0480 | ||
PDC-PTX1 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [11] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
60%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 5 µM | ||||
| Evaluation Method | MTT assay | ||||
| Description |
Antiproliferative results showed that PTX1 inhibited cell proliferation by 18.7%. The anti-proliferative activity of CPT1 was diminished by 1.9-fold as compared to CPT whereas the activity of CPT2 was comparable to CPT, since CPT2 reduced the cell viability to 61%.
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [11] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
80%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 5 µM | ||||
| Evaluation Method | MTT assay | ||||
| Description |
The cytotoxicity of PTX and PTX1 was further evaluated in the normal human embryonic kidney cells (HEK-293) at 5 uM which showed reduced cell proliferation by ~34% and 18%, respectively, after 72 h using MTT assay, as shown in Figure 2.
|
||||
| In Vitro Model | Normal | HEK-298 cell | Homo sapiens | ||
AEZS-108 [Terminated in Phase 3]
Identified from the Human Clinical Data
| Experiment 1 Reporting the Activity Data of This PDC | [12] | ||||
| Indication | Castration and taxane resistant prostate cancer | ||||
| Efficacy Data | Vomit |
32%
|
|||
| Description |
The most common all-grade adverse events were hematologic (22; 88%), fatigue (19; 76%), anorexia (13; 52%),alopecia(13; 52%), nausea (13; 52%), and vomiting (8;32%).
|
||||
| In Vivo Model | Men with histologically confirmed prostatic adenocarcinoma. | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [12] | ||||
| Indication | Castration and taxane resistant prostate cancer | ||||
| Efficacy Data | Neutropenia |
56%
|
|||
| Description |
The most common hematologic adverse event was neutropenia at 56% (all grades).
|
||||
| In Vivo Model | Men with histologically confirmed prostatic adenocarcinoma. | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [12] | ||||
| Indication | Castration and taxane resistant prostate cancer | ||||
| Efficacy Data | Nausea toxicity |
52%
|
|||
| Description |
The most common all-grade adverse events were hematologic (22; 88%), fatigue (19; 76%), anorexia (13; 52%),alopecia(13; 52%), nausea (13; 52%), and vomiting (8;32%).
|
||||
| In Vivo Model | Men with histologically confirmed prostatic adenocarcinoma. | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [12] | ||||
| Indication | Castration and taxane resistant prostate cancer | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
3.8 months
|
|||
| Description |
With a median follow-up of 16.1 months (range, 3.2-36.1), the median PFS was 3.8 months (95% confidence interval [CI], 2.1-4.4) and median OS was 6.0 months (95% CI, 4.2-10.1; Figure 3).
|
||||
| In Vivo Model | Men with histologically confirmed prostatic adenocarcinoma. | ||||
| Experiment 5 Reporting the Activity Data of This PDC | [12] | ||||
| Indication | Castration and taxane resistant prostate cancer | ||||
| Efficacy Data | Median overall survival (mOS) |
6.0 months
|
|||
| Description |
With a median follow-up of 16.1 months (range, 3.2-36.1), the median PFS was 3.8 months (95% confidence interval [CI], 2.1-4.4) and median OS was 6.0 months (95% CI, 4.2-10.1; Figure 3).
|
||||
| In Vivo Model | Men with histologically confirmed prostatic adenocarcinoma. | ||||
| Experiment 6 Reporting the Activity Data of This PDC | [12] | ||||
| Indication | Castration and taxane resistant prostate cancer | ||||
| Efficacy Data | Hematologic toxicity |
88%
|
|||
| Description |
The most common all-grade adverse events were hematologic (22; 88%), fatigue (19; 76%), anorexia (13; 52%),alopecia(13; 52%), nausea (13; 52%), and vomiting (8;32%).
|
||||
| In Vivo Model | Men with histologically confirmed prostatic adenocarcinoma. | ||||
| Experiment 7 Reporting the Activity Data of This PDC | [12] | ||||
| Indication | Castration and taxane resistant prostate cancer | ||||
| Efficacy Data | Grade 3/4 nonhematologic toxicities |
24%
|
|||
| Description |
Six of 25 patients (24%) experienced Grade 3 or 4 nonhematologic toxicities
|
||||
| In Vivo Model | Men with histologically confirmed prostatic adenocarcinoma. | ||||
| Experiment 8 Reporting the Activity Data of This PDC | [12] | ||||
| Indication | Castration and taxane resistant prostate cancer | ||||
| Efficacy Data | Grade ≥ 3 hematologic toxicity |
56%
|
|||
| Description |
Fourteen of 25 patients (56%) experienced a Grade ≥ 3 hematologic toxicity
|
||||
| In Vivo Model | Men with histologically confirmed prostatic adenocarcinoma. | ||||
| Experiment 9 Reporting the Activity Data of This PDC | [12] | ||||
| Indication | Castration and taxane resistant prostate cancer | ||||
| Efficacy Data | Fatigue |
76%
|
|||
| Description |
The most common all-grade adverse events were hematologic (22; 88%), fatigue (19; 76%), anorexia (13; 52%),alopecia(13; 52%), nausea (13; 52%), and vomiting (8;32%).
|
||||
| In Vivo Model | Men with histologically confirmed prostatic adenocarcinoma. | ||||
| Experiment 10 Reporting the Activity Data of This PDC | [12] | ||||
| Indication | Castration and taxane resistant prostate cancer | ||||
| Efficacy Data | Alopecia toxicity |
52.00%
|
|||
| Description |
The most common all-grade adverse events were hematologic (22; 88%), fatigue (19; 76%), anorexia (13; 52%),alopecia(13; 52%), nausea (13; 52%), and vomiting (8;32%).
|
||||
| In Vivo Model | Men with histologically confirmed prostatic adenocarcinoma. | ||||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Uveal melanoma | ||||
| Efficacy Data | Cell viability |
0%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 5 µM | ||||
| Evaluation Method | MTT assay | ||||
| Description |
OCM3DOX320cells did not show significant difference in cell viability in the presence of 1 uM DOX when compared to untreated control cells. 1 uM DOX induced significant cell death of OCM3 cells but did not cause significant cell death in OCM3DOX320cells, confirming DOX resistance. AN-152 at lower concentration (40 nM, 320 nM) increased cell proliferation significantly compared to equimolar dose of DOX which does not have any effect on cell viability at this concentration in OCM3 cells. However, higher concentrations of AN-152 can effectively inhibit cell proliferation in both cell lines. Higher concentrations (1-5 uM) of DOX and AN-152 showed no significantly different effect either on OCM3 or OCM3DOX320cells (Fig. 3).
Click to Show/Hide
|
||||
| In Vitro Model | Cutaneous melanoma | OCM-3 cell | CVCL_6937 | ||
| Half life period | 2 h | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Uveal melanoma | ||||
| Efficacy Data | Cell viability |
5%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 1 µM | ||||
| Evaluation Method | MTT assay | ||||
| Description |
OCM3DOX320cells did not show significant difference in cell viability in the presence of 1 uM DOX when compared to untreated control cells. 1 uM DOX induced significant cell death of OCM3 cells but did not cause significant cell death in OCM3DOX320cells, confirming DOX resistance. AN-152 at lower concentration (40 nM, 320 nM) increased cell proliferation significantly compared to equimolar dose of DOX which does not have any effect on cell viability at this concentration in OCM3 cells. However, higher concentrations of AN-152 can effectively inhibit cell proliferation in both cell lines. Higher concentrations (1-5 uM) of DOX and AN-152 showed no significantly different effect either on OCM3 or OCM3DOX320cells (Fig. 3).
Click to Show/Hide
|
||||
| In Vitro Model | Cutaneous melanoma | OCM-3 cell | CVCL_6937 | ||
| Half life period | 2 h | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Uveal melanoma | ||||
| Efficacy Data | Cell viability |
6%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 2.5 µM | ||||
| Evaluation Method | MTT assay | ||||
| Description |
OCM3DOX320cells did not show significant difference in cell viability in the presence of 1 uM DOX when compared to untreated control cells. 1 uM DOX induced significant cell death of OCM3 cells but did not cause significant cell death in OCM3DOX320cells, confirming DOX resistance. AN-152 at lower concentration (40 nM, 320 nM) increased cell proliferation significantly compared to equimolar dose of DOX which does not have any effect on cell viability at this concentration in OCM3 cells. However, higher concentrations of AN-152 can effectively inhibit cell proliferation in both cell lines. Higher concentrations (1-5 uM) of DOX and AN-152 showed no significantly different effect either on OCM3 or OCM3DOX320cells (Fig. 3).
Click to Show/Hide
|
||||
| In Vitro Model | Cutaneous melanoma | OCM-3 cell | CVCL_6937 | ||
| Half life period | 2 h | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Uveal melanoma | ||||
| Efficacy Data | Cell viability |
25%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 5 µM | ||||
| Evaluation Method | MTT assay | ||||
| Description |
OCM3DOX320cells did not show significant difference in cell viability in the presence of 1 uM DOX when compared to untreated control cells. 1 uM DOX induced significant cell death of OCM3 cells but did not cause significant cell death in OCM3DOX320cells, confirming DOX resistance. AN-152 at lower concentration (40 nM, 320 nM) increased cell proliferation significantly compared to equimolar dose of DOX which does not have any effect on cell viability at this concentration in OCM3 cells. However, higher concentrations of AN-152 can effectively inhibit cell proliferation in both cell lines. Higher concentrations (1-5 uM) of DOX and AN-152 showed no significantly different effect either on OCM3 or OCM3DOX320cells (Fig. 3).
Click to Show/Hide
|
||||
| In Vitro Model | Cutaneous melanoma | OCM3DOX320 cell | CVCL_6937 | ||
| Half life period | 2 h | ||||
| Experiment 5 Reporting the Activity Data of This PDC | [14] | ||||
| Indication | Uveal melanoma | ||||
| Efficacy Data | Cell viability |
36.30%
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 5 µM | ||||
| Evaluation Method | MTS assay | ||||
| MOA of PDC |
Our results show that AEZS-108 upregulates the expression of MASPIN/SERPINB5 tumor suppressor gene, which is downregulated in normal uvea and UM specimens independently from the LHRH receptor-ligand interaction. AEZS-108 also substantially downregulates hypoxia-inducible factor 1 alpha (HIF1A) expression.
|
||||
| Description |
In order to investigate whether AEZS-108 inhibits cell proliferation and its extent, OCM3 cells were treated either with 5 M AEZS-108 or equal amount of doxorubicin. MTS assay was performed after 24 and 48 hours of treatment. AEZS-108 and doxorubicin have been shown to reduce cell proliferation by 36.3% (p< 0.001) and 62.9% (p< 0.001) respectively after 24 hours, and by 84.7% (p< 0.001) and 89.7% (p< 0.001) respectively after 48 hours, (Figure 2).
Click to Show/Hide
|
||||
| In Vitro Model | Cutaneous melanoma | OCM-3 cell | CVCL_6937 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Uveal melanoma | ||||
| Efficacy Data | Cell viability |
48%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 2.5 µM | ||||
| Evaluation Method | MTT assay | ||||
| Description |
OCM3DOX320cells did not show significant difference in cell viability in the presence of 1 uM DOX when compared to untreated control cells. 1 uM DOX induced significant cell death of OCM3 cells but did not cause significant cell death in OCM3DOX320cells, confirming DOX resistance. AN-152 at lower concentration (40 nM, 320 nM) increased cell proliferation significantly compared to equimolar dose of DOX which does not have any effect on cell viability at this concentration in OCM3 cells. However, higher concentrations of AN-152 can effectively inhibit cell proliferation in both cell lines. Higher concentrations (1-5 uM) of DOX and AN-152 showed no significantly different effect either on OCM3 or OCM3DOX320cells (Fig. 3).
Click to Show/Hide
|
||||
| In Vitro Model | Cutaneous melanoma | OCM3DOX320 cell | CVCL_6937 | ||
| Half life period | 2 h | ||||
| Experiment 7 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Uveal melanoma | ||||
| Efficacy Data | Cell viability |
80%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 1 µM | ||||
| Evaluation Method | MTT assay | ||||
| Description |
OCM3DOX320cells did not show significant difference in cell viability in the presence of 1 uM DOX when compared to untreated control cells. 1 uM DOX induced significant cell death of OCM3 cells but did not cause significant cell death in OCM3DOX320cells, confirming DOX resistance. AN-152 at lower concentration (40 nM, 320 nM) increased cell proliferation significantly compared to equimolar dose of DOX which does not have any effect on cell viability at this concentration in OCM3 cells. However, higher concentrations of AN-152 can effectively inhibit cell proliferation in both cell lines. Higher concentrations (1-5 uM) of DOX and AN-152 showed no significantly different effect either on OCM3 or OCM3DOX320cells (Fig. 3).
Click to Show/Hide
|
||||
| In Vitro Model | Cutaneous melanoma | OCM3DOX320 cell | CVCL_6937 | ||
| Half life period | 2 h | ||||
| Experiment 8 Reporting the Activity Data of This PDC | [14] | ||||
| Indication | Uveal melanoma | ||||
| Efficacy Data | Cell viability |
84.70%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 5 µM | ||||
| Evaluation Method | MTS assay | ||||
| MOA of PDC |
Our results show that AEZS-108 upregulates the expression of MASPIN/SERPINB5 tumor suppressor gene, which is downregulated in normal uvea and UM specimens independently from the LHRH receptor-ligand interaction. AEZS-108 also substantially downregulates hypoxia-inducible factor 1 alpha (HIF1A) expression.
|
||||
| Description |
In order to investigate whether AEZS-108 inhibits cell proliferation and its extent, OCM3 cells were treated either with 5 M AEZS-108 or equal amount of doxorubicin. MTS assay was performed after 24 and 48 hours of treatment. AEZS-108 and doxorubicin have been shown to reduce cell proliferation by 36.3% (p< 0.001) and 62.9% (p< 0.001) respectively after 24 hours, and by 84.7% (p< 0.001) and 89.7% (p< 0.001) respectively after 48 hours, (Figure 2).
Click to Show/Hide
|
||||
| In Vitro Model | Cutaneous melanoma | OCM-3 cell | CVCL_6937 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Uveal melanoma | ||||
| Efficacy Data | Cell viability |
105%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 320 nM | ||||
| Evaluation Method | MTT assay | ||||
| Description |
OCM3DOX320cells did not show significant difference in cell viability in the presence of 1 uM DOX when compared to untreated control cells. 1 uM DOX induced significant cell death of OCM3 cells but did not cause significant cell death in OCM3DOX320cells, confirming DOX resistance. AN-152 at lower concentration (40 nM, 320 nM) increased cell proliferation significantly compared to equimolar dose of DOX which does not have any effect on cell viability at this concentration in OCM3 cells. However, higher concentrations of AN-152 can effectively inhibit cell proliferation in both cell lines. Higher concentrations (1-5 uM) of DOX and AN-152 showed no significantly different effect either on OCM3 or OCM3DOX320cells (Fig. 3).
Click to Show/Hide
|
||||
| In Vitro Model | Cutaneous melanoma | OCM3DOX320 cell | CVCL_6937 | ||
| Half life period | 2 h | ||||
| Experiment 10 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Uveal melanoma | ||||
| Efficacy Data | Cell viability |
120%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 40 nM | ||||
| Evaluation Method | MTT assay | ||||
| Description |
OCM3DOX320cells did not show significant difference in cell viability in the presence of 1 uM DOX when compared to untreated control cells. 1 uM DOX induced significant cell death of OCM3 cells but did not cause significant cell death in OCM3DOX320cells, confirming DOX resistance. AN-152 at lower concentration (40 nM, 320 nM) increased cell proliferation significantly compared to equimolar dose of DOX which does not have any effect on cell viability at this concentration in OCM3 cells. However, higher concentrations of AN-152 can effectively inhibit cell proliferation in both cell lines. Higher concentrations (1-5 uM) of DOX and AN-152 showed no significantly different effect either on OCM3 or OCM3DOX320cells (Fig. 3).
Click to Show/Hide
|
||||
| In Vitro Model | Cutaneous melanoma | OCM3DOX320 cell | CVCL_6937 | ||
| Half life period | 2 h | ||||
| Experiment 11 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Uveal melanoma | ||||
| Efficacy Data | Cell viability |
130%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 40 nM | ||||
| Evaluation Method | MTT assay | ||||
| Description |
OCM3DOX320cells did not show significant difference in cell viability in the presence of 1 uM DOX when compared to untreated control cells. 1 uM DOX induced significant cell death of OCM3 cells but did not cause significant cell death in OCM3DOX320cells, confirming DOX resistance. AN-152 at lower concentration (40 nM, 320 nM) increased cell proliferation significantly compared to equimolar dose of DOX which does not have any effect on cell viability at this concentration in OCM3 cells. However, higher concentrations of AN-152 can effectively inhibit cell proliferation in both cell lines. Higher concentrations (1-5 uM) of DOX and AN-152 showed no significantly different effect either on OCM3 or OCM3DOX320cells (Fig. 3).
Click to Show/Hide
|
||||
| In Vitro Model | Cutaneous melanoma | OCM-3 cell | CVCL_6937 | ||
| Half life period | 2 h | ||||
References