
Drug Information
General Information of This Drug
| Drug ID | DRG00012 | |||||
|---|---|---|---|---|---|---|
| Drug Name | Paclitaxel | |||||
| Synonyms |
33069-62-4; P88XT4IS4D; Paclitaxel; Taxol; Taxol A; Yewtaxan; Genaxol; Plaxicel; Abraxane; Ebetaxel; Genetaxyl; Capxol; Paxene; Onxol; Cyclopax; Genexol; Intaxel; Mitotax; TaxAlbin; OncoGel; Pacliex; Paxceed; EmPAC; Onxal; Zisu; Taxus stent; Taxus Liberte; ABI-007; Padexol; EndoTAG 1; LipoPac; Tocosol Paclitaxel; (-)-Paclitaxel; Nanoxel; Paclitaxol; Sindaxel; NSC-125973; Coroflex Please; Cypher select; Taxus Express; LEP-ETU; Genexol-PM; (NAB)-Paclitaxel; MBT 0206; Infinnium; Taxus; HSDB 6839; ABI 007; DHP 107; DHP-107; Abraxane I.V. Suspension; BMS 181339-01; BMS-181339-01; UNII-P88XT4IS4D; DRG-0190; Paclitaxel (Taxol); NK 105; NSC125973; Paclitaxel (taxus canadensis); QW 8184; CCRIS 8143; Liposome-entrapped paclitaxel easy-to-use; DTXSID9023413; CHEBI:45863; ABI-007 COMPONENT PACLITAXEL; IG 001; MFCD00869953; NK-105; 5beta,20-Epoxy-1,2-alpha,4,7beta,10beta,13alpha-hexahydroxytax-11-en-9-one 4,10-diacetate 2-benzoate 13-ester with (2R,3S)-N-benzoyl-3-phenylisoserine; QW-8184; CHEMBL428647; DTXCID603413; (2aR,4S,4aS,6R,9S,11S,12S,12aR,12bS)-9-(((2R,3S)-3-benzamido-2-hydroxy-3-phenylpropanoyl)oxy)-12-(benzoyloxy)-4,11-dihydroxy-4a,8,13,13-tetramethyl-5-oxo-2a,3,4,4a,5,6,9,10,11,12,12a,12b-dodecahydro-1H-7,11-methanocyclodeca[3,4]benzo[1,2-b]oxete-6,12b-diyl diacetate; nab-paclitaxel; ORAXOL COMPONENT PACLITAXEL; Paclitaxel [USAN:USP:INN:BAN]; Abraxane (albumin-bound suspension); ABRAXANE COMPONENT PACLITAXEL; MBT-0206; ABI 007 COMPONENT PACLITAXEL; (2aR-(2aalpha,4beta,4abeta,6beta,9alpha(alpha R*,betaS*),11alpha,12alpha,12balpha))-beta-(Benzoylamino)-alpha-hydroxybenzenepropanoic acid 6,12b-bis(acetyloxy)-12-(benzoyloxy)-2a,3,4,4a,5,6,9,10,11,12,12a,12b-dodecahydro-4,11-dihydroxy-4a,8,13,13-tetramethyl-5-oxo-7,11-methano-1H-cyclodeca(3,4)benz(1,2-b)oxet-9-yl ester; NCGC00164367-01; NAB-PACLITAXEL COMPONENT PACLITAXEL; 7,11-Methano-1H-cyclodeca[3,4]benz[1,2-b]oxete, benzenepropanoic acid deriv.; NSC 125973; PACLITAXEL (MART.); PACLITAXEL [MART.]; PACLITAXEL (USP-RS); PACLITAXEL [USP-RS]; (1S,2S,3R,4S,7R,9S,10S,12R,15S)-4,12-bis(acetyloxy)-1,9-dihydroxy-15-{[(2R,3S)-2-hydroxy-3-phenyl-3-(phenylformamido)propanoyl]oxy}-10,14,17,17-tetramethyl-11-oxo-6-oxatetracyclo[11.3.1.0^{3,10}.0^{4,7}]heptadec-13-en-2-yl benzoate; PACLITAXEL (EP MONOGRAPH); PACLITAXEL (USP IMPURITY); PACLITAXEL [EP MONOGRAPH]; PACLITAXEL [USP IMPURITY]; Anzatax; Cynviloq; PACLITAXEL (USP MONOGRAPH); PACLITAXEL [USP MONOGRAPH]; Xorane; Paclitaxel (USAN:USP:INN:BAN); Bris Taxol; Taxol, Bris; SMR000857385; EndoTAG-1; SR-01000075350; paclitaxelum; Nanotaxel; Paclical; Pacligel; Paxoral; Paclitaxel?; Paclitaxel,(S); Abraxane (TN); (2alpha,5beta,7beta,10beta,13alpha)-4,10-bis(acetyloxy)-1,7-dihydroxy-13-({(2R,3S)-2-hydroxy-3-phenyl-3-[(phenylcarbonyl)amino]propanoyl}oxy)-9-oxo-5,20-epoxytax-11-en-2-yl benzoate; [diacetoxy-[(2R,3S)-3-benzamido-2-hydroxy-3-phenyl-propanoyl]oxy-dihydroxy-tetramethyl-oxo-[?]yl] benzoate; 4alpha,10beta-bis(acetyloxy)-13alpha-((2S,3S)-3-benzamido-2-hydroxy-3-phenylpropanoyloxy)-1,7beta-dihydroxy-9-oxo-5beta,20-epoxytax-11-en-2alpha-yl benzoate; 4alpha,10beta-bis(acetyloxy)-13alpha-[(2S,3S)-3-benzamido-2-hydroxy-3-phenylpropanoyloxy]-1,7beta-dihydroxy-9-oxo-5beta,20-epoxytax-11-en-2alpha-yl benzoate; Paclitaxel; 5beta,20-Epoxy-1,7beta-dihydroxy-9-oxotax-11-ene-2alpha,4,10beta,13alpha-tetrayl 4,10-diacetate 2-benzoate 13-[(2R,3S)-3-(benzoylamino)-2-hydroxy-3-phenylpropanoate]; Taxol; Docetaxel Anhydrous Impurity F; Docetaxel Impurity F; Taxol (Paclitaxel); CAS-33069-62-4; BMS-181339; Paclitaxel-SSMM-VIP; P-SSMM-VIP; PACLITAXEL [MI]; PACLITAXEL [INN]; PACLITAXEL [JAN]; Prestwick3_000155; PACLITAXEL [HSDB]; PACLITAXEL [USAN]; PACLITAXELPACLITAXEL; TAXOL (TN); PACLITAXEL [VANDF]; SCHEMBL3976; 3PPC5TL76P; Nova-12005; PACLITAXEL [WHO-DD]; Paclitaxel, Taxus brevifolia; BIDD:PXR0046; BSPBio_000290; KBioGR_002509; KBioSS_002517; Paclitaxel (JAN/USP/INN); MLS002154218; MLS002695976; OAS-PAC-100; PACLITAXEL [EMA EPAR]; BPBio1_000320; GTPL2770; MEGxp0_001940; Taxol (TN) (Bristol Meyers); PACLITAXEL [GREEN BOOK]; PACLITAXEL [ORANGE BOOK]; ACon1_002231; KBio2_002509; KBio2_005077; KBio2_007645; KBio3_002987; ANX-513; DHP-208; DTS-301; L01CD01; SDP-013; cMAP_000068; HMS2090D07; HMS2095O12; HMS2231A16; HMS3712O12; HY-B0015; MPI-5018; Tox21_112107; BDBM50001839; NSC745099; AKOS007930675; AKOS015969673; AKOS025312303; CCG-220155; CS-1145; DB01229; GS-6554; NSC-745099; NCGC00164367-02; NCGC00164367-03; NCGC00164367-04; NCGC00164367-05; NCGC00164367-10; Paclitaxel, From Taxus brevifolia, 95%; (2aR,4S,4aS,6R,9S,11S,12S,12aR,12bS)-1,2a,3,4,4a,6,9,10,11,12,12a,12b-Dodecahydro-4,6,9,11,12,12b-hexahydroxy-4a,8,13,13-tetramethyl-7,11-methano-5H-cyclodeca(3,4)benz(1,2-b)oxet-5-one 6,12b-diacetate, 12-benzoate, 9-ester with (2R,3S)-N-benzoyl-3-phenylisoserine; NCI60_000601; Paclitaxel, from Taxus yannanensis, powder; 1ST000431; PACLITAXEL IMPURITY L [EP IMPURITY]; AB00513812; D00491; EN300-117275; M02242; N88686; AB00513812-02; AB00513812-03; Paclitaxel, Antibiotic for Culture Media Use Only; Q423762; 7,4]benz[1,2-b]oxete,benzenepropanoic acid deriv.; Q-201533; SR-01000075350-1; SR-01000075350-3; SR-01000075350-6; SR-01000075350-7; SR-01000075350-9; BRD-K62008436-001-03-1; BRD-K62008436-001-05-6; BRD-K62008436-001-22-1; Paclitaxel, from semisynthetic (from Taxus sp.), >=97%; Paclitaxel, European Pharmacopoeia (EP) Reference Standard; Paclitaxel, from Taxus brevifolia, >=95% (HPLC), powder; Paclitaxel, United States Pharmacopeia (USP) Reference Standard; 12-benzoate, 9-ester with (2R,3S)-N-benzoyl-3-phenylisoserine; Paclitaxel protein-bound particles for injectable suspension (albumin-bound); Paclitaxel, Pharmaceutical Secondary Standard; Certified Reference Material; Paclitaxel natural for peak identification, European Pharmacopoeia (EP) Reference Standard; (1S,2S,3R,4S,5R,7S,8S,10R,13S)-4,10-Diacetoxy-2-benzoyloxy-5,20-epoxy-1,7-dihydroxy-9-oxotax-11-en-13-yl (2R,3S)-3-benzoylamino-2-hydroxy-3-phenylpropionate; (2aR,4S,4aS,6R,9S,11S,12S,12aR,12bS)-1,2a,3,4,4a,6,9,10,11,12,12a,12b-Dodecahydro 4,6,9,11,12,12b-hexahydroxy-4a,8,13,13-tetramethyl-7,11-methano 5Hcyclodeca(3,4)benz(1,2-b)oxet-5-one 6,12b-diacetate,; (2aR,4S,4aS,6R,9S,11S,12S,12aR,12bS)-4,6,12b-Tris(acetyloxy)-12-(benzoyloxy)-2a,3,4,4a,5,6,9,10,11,12,12a,12b-dodecahydro-11-hydroxy-4a,8,13,13-tetramethyl-5-oxo-7,11-methano-1H-cyclodeca[3,4]benz[1,2-b]oxet-9-yl (alphaR,betaS)-beta-(benzoylamino)-alpha-hydroxybenzenepropanoate; (2aR,4S,4aS,6R,9S,11S,12S,12aR,12bS)-6,12b-bis(acetyloxy)-12-(benzoyloxy)-2a,3,4,4a,5,6,9,10,11,12,12a,12b-dodecahydro-4,11-dihydroxy-4a,8,13,13-tetramethyl-5-oxo-7,11-methano-1H-cyclodeca[3,4]benz[1,2-b]oxet-9-yl (aR,bS)-b-(benzoylamino)-a-hydroxybenzenepropanoate; (2aR,4S,4aS,6R,9S,11S,12S,12aR,12bS)-9-(((2R,3S)-3-benzamido-2-hydroxy-3-phenylpropanoyl)oxy)-12-(benzoyloxy)-4,11-dihydroxy-4a,8,13,13-tetramethyl-5-oxo-3,4,4a,5,6,9,10,11,12,12a-decahydro-1H-7,11-methanocyclodeca[3,4]benzo[1,2-b]oxete-6,12b(2aH)-diyl diacetate; (2aR-(2aalpha,4beta,4abeta,6beta,9alpha(alpha R*,betaS*),11alpha,12alpha,12balpha))-beta-(Benzoylamino)-alpha-hydroxybenzenepropanoic acid 6,12b-bis(acetyloxy)-12-(benzoyloxy)-2a,3,4,4a,5,6,9,10,11,12; (2beta,5beta,7alpha,8alpha,10alpha,13alpha)-4,10-bis(acetyloxy)-1,7-dihydroxy-13-({(2R,3S)-2-hydroxy-3-phenyl-3-[(phenylcarbonyl)amino]propanoyl}oxy)-9-oxo-5,20-epoxytax-11-en-2-yl benzoate; ,12a,12b-dodecahydro-4,11-dihydroxy-4a,8,13,13-tetramethyl-5-oxo-7,11-methano-1H-cyclodeca(3,4)benz(1,2-b)oxet-9-yl ester; ,13-tetramethyl-5-oxo-7,11-methano-1H-cyclodeca[3,4]benz[1,2-b]oxet-9-yl ester, (alphaR,betaS)- (9CI); -cyclodeca[3,4]benz[1,2-b]oxet-9-yl ester, [2aR-[2aalpha,4beta,4abeta,6beta,9alpha(aR*,betaS*),11alpha,12alpha,12aalpha,12balpha]]-; [(1S,2S,3R,4S,7R,9S,10S,12R,15S)-4,12-diacetyloxy-15-[(2R,3S)-3-benzamido-2-hydroxy-3-phenylpropanoyl]oxy-1,9-dihydroxy-10,14,17,17-tetramethyl-11-oxo-6-oxatetracyclo[11.3.1.03,10.04,7]heptadec-13-en-2-yl] benzoate; [(1S,2S,3R,4S,7R,9S,10S,12R,15S)-4,12-diacetyloxy-15-[(2R,3S)-3-benzamido-2-hydroxy-3-phenylpropanoyl]oxy-1,9-dihydroxy-10,14,17,17-tetramethyl-11-oxo-6-oxatetracyclo[11.3.1.03,10.04,7]heptadec-13-en-2-yl]benzoate; 1203669-79-7; 4,7beta,10beta-tris(acetyloxy)-13alpha-[[(2R,3S)-3-benzamido-2-hydroxy-3-phenylpropanoyl]oxy]-1-hydroxy-9-oxo-5beta,20-epoxytax-11-en-2alpha-yl benzoate; 5-BETA,20-EPOXY-1,2-ALPHA,4,7-BETA,10-BETA,13-ALPHA-HEXAHYDROXY-TAX-11-EN-9-ONE 4,10-DIACETATE 2-BENZOATE 13-ESTER WITH (2R,3S)-N-BENZOYL-3-PHENYL-ISOSERINE; 5beta,20-Epoxy-1,2 alpha, 4,7beta, 10beta, 13alpha-hexahydroxy tax-11-en-9-one 4,10-diacetate 2-benzoate 13-ester with (2R, 3S)-N-benzoyl-3-phenylisoserine; BENZENEPROPANOIC ACID, .BETA.-(BENZOYLAMINO)-.ALPHA.-HYDROXY-, (2AR,4S,4AS,6R,9S,11S,12S,12AR,12BS)-6,12B-BIS(ACETYLOXY)-12-(BENZOYLOXY)-2A,3,4,4A,5,6,9,10,11,12,12A,12B-DODECAHYDRO-4,11-DIHYDROXY-4A,8,13,13-TETRAMETHYL-5-OXO-7,11-METHANO-1H-CYCLODECA(3,4)BENZ(1,2-B)OXET-9-YL ESTER, (.ALPHA.R,.BETA.S)-; Benzenepropanoic acid, 6,12b-bis(acetyl oxy)-12-(benzoyloxy)- 2a,3,4,4a,5,6,9,10,11,12,12a,12b,- dodecahydro-4,11- dihydroxy-4a,8,13,13-tetramethyl-5-oxo- 7,11-methano- 1H-cyclodeca[3,4]benz[1,2-b]oxet-9-yl ester, [2aR- [2a.alpha.,4.beta.,4a.beta.,6.beta.,9.alpha.(alpha. R*,.beta.S*),11.alpha.,12.alpha.,12a.alpha.,12b.alpha.]]-; Benzenepropanoic acid, b-(benzoylamino)-.alpha.-hydroxy-, (2aR,4S,4aS,6R,9S,11S,12S,12aR,12bS)-6,12b-bis(acetyloxy)-12-(benzoyloxy)-2a,3,4,4a,5,6,9,10,11,12,12a,12b-dodecahydro-4,11-dihydroxy-4a,8,13,13-tetramethyl-5-oxo-7,11-methano-1H-cyclodeca[3,4]benz[1,2-b]oxet-9-yl ester, (aR,bS)-; Benzenepropanoic acid, beta-(benzoylamino)-alpha-hydroxy-, (2aR,4S,4aS,6R,9S,11S,12S,12aR,12bS)-4,6,12b-tris(acetyloxy)-12-(benzoyloxy)-2a,3,4,4a,5,6,9,10,11,12,12a,12b-dodecahydro-11-hydroxy-4a,8,13,13-tetramethyl-5-oxo-7,11-methano-1H-cyclodeca[3,4]benz[1,2-b]oxet-9-yl ester, (alphaR,betaS)-; Benzenepropanoic acid, beta-(benzoylamino)-alpha-hydroxy-, (2aR,4S,4aS,6R,9S,11S,12S,12aR,12bS)-6,12b-bis(acetyloxy)-12-(benzoyloxy)-2a,3,4,4a,5,6,9,10,11,12,12a,12b-dodecahydro-4,11-dihydroxy-4a,8,13; Benzenepropanoic acid, beta-(benzoylamino)-alpha-hydroxy-, (2aR,4S,4aS,6R,9S,11S,12S,12aR,12bS)-6,12b-bis(acetyloxy)-12-(benzoyloxy)-2a,3,4,4a,5,6,9,10
Click to Show/Hide
|
|||||
| Target(s) | Microtubule (MT) | Target Info | ||||
| Structure |
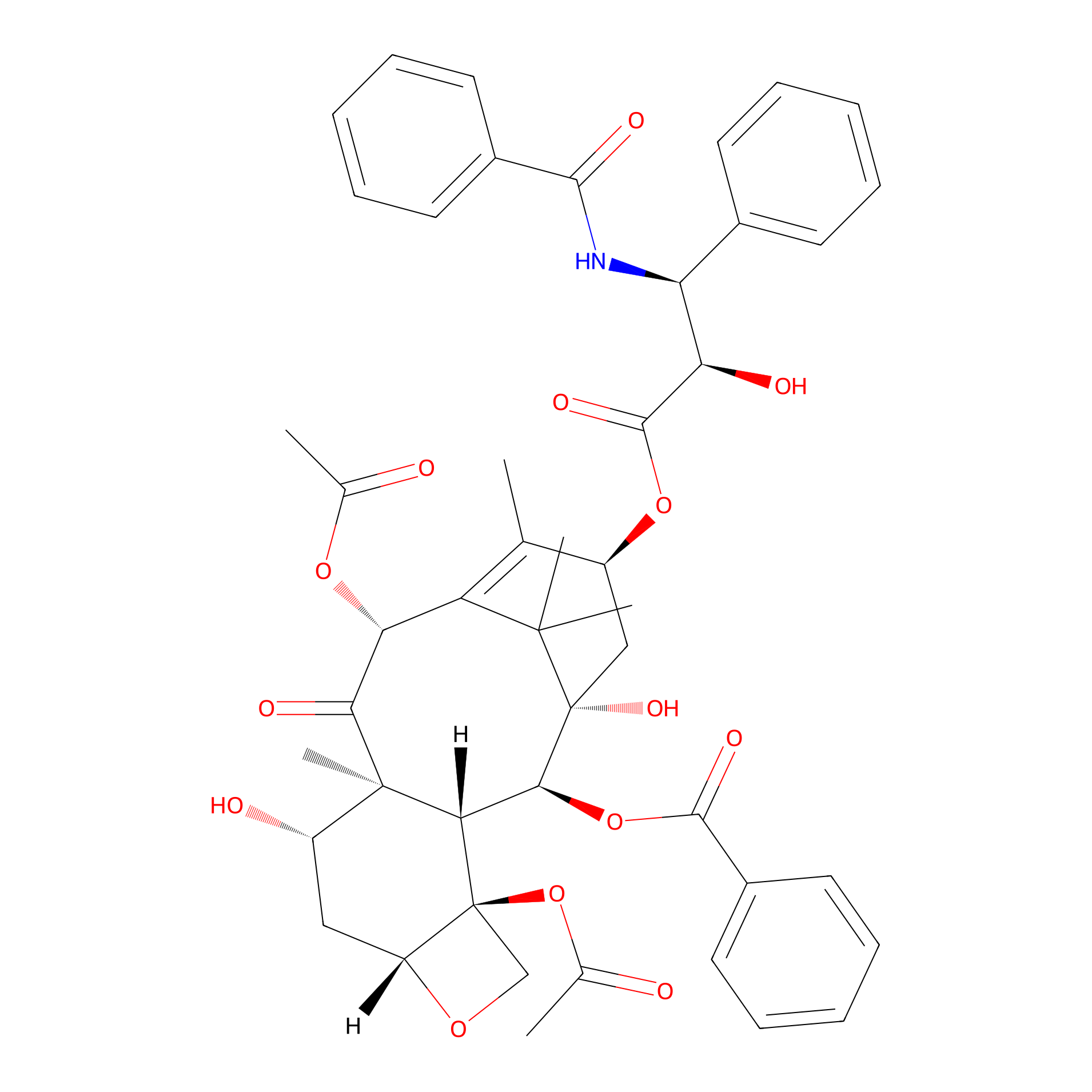
|
|||||
| Formula |
C47H51NO14
|
|||||
| #Ro5 Violations (Lipinski): 3 | Molecular Weight (mw) | 853.9 | ||||
| Lipid-water partition coefficient (xlogp) | 2.5 | |||||
| Hydrogen Bond Donor Count (hbonddonor) | 4 | |||||
| Hydrogen Bond Acceptor Count (hbondacc) | 14 | |||||
| Rotatable Bond Count (rotbonds) | 14 | |||||
| PubChem CID | ||||||
| Canonical smiles |
CC1=C2C(C(=O)C3(C(CC4C(C3C(C(C2(C)C)(CC1OC(=O)C(C(C5=CC=CC=C5)NC(=O)C6=CC=CC=C6)O)O)OC(=O)C7=CC=CC=C7)(CO4)OC(=O)C)O)C)OC(=O)C
|
|||||
| InChI |
InChI=1S/C47H51NO14/c1-25-31(60-43(56)36(52)35(28-16-10-7-11-17-28)48-41(54)29-18-12-8-13-19-29)23-47(57)40(61-42(55)30-20-14-9-15-21-30)38-45(6,32(51)22-33-46(38,24-58-33)62-27(3)50)39(53)37(59-26(2)49)34(25)44(47,4)5/h7-21,31-33,35-38,40,51-52,57H,22-24H2,1-6H3,(H,48,54)/t31-,32-,33+,35-,36+,37+,38-,40-,45+,46-,47+/m0/s1
|
|||||
| InChIKey |
RCINICONZNJXQF-MZXODVADSA-N
|
|||||
| IUPAC Name |
[(1S,2S,3R,4S,7R,9S,10S,12R,15S)-4,12-diacetyloxy-15-[(2R,3S)-3-benzamido-2-hydroxy-3-phenylpropanoyl]oxy-1,9-dihydroxy-10,14,17,17-tetramethyl-11-oxo-6-oxatetracyclo[11.3.1.03,10.04,7]heptadec-13-en-2-yl] benzoate
|
|||||
The activity data of This Drug
| Standard Type | Value | Administration times | Administration dosage | Cell line | Cell line ID | Ref. |
|---|---|---|---|---|---|---|
| Cell viability | 75% | 24 h | 200 nM | U-87MG cell | CVCL_0022 | [1] |
| Median lethal dose (LD50) | 35 mg/kg | 24 h | 200 nM | U-87MG cell | CVCL_0022 | [1] |
| Percent survival | 0% | 20 days | 17.3 mg/kg | MDA-MB-231 cell | CVCL_0062 | [1] |
| Percent survival | 0% | 25 days | 17.3 mg/kg | U87MG-Luc cell | CVCL_5J15 | [1] |
| Tumor Growth Inhibition value (TGI) | 28.47% | 16 days | 4 mg/kg | 4T1-mCherry-Luc cell | CVCL_C8UZ | [2] |
| Half Maximal Effective Concentration (EC50) | 12.25±0.13 nM | 24 h | N.A. | U-87MG cell | CVCL_0022 | [3] |
| Half Maximal Effective Concentration (EC50) | 41.3±1.5 nM | 24 h | N.A. | U87MG-PR cell | CVCL_0022 | [3] |
| Half Maximal Inhibitory Concentration (IC50) | 0.006 µg/mL | 72 h | N.A. | HCT 116 cell | CVCL_0291 | [4] |
| Half Maximal Inhibitory Concentration (IC50) | 0.01 µg/mL | 72 h | N.A. | 4T1 cell | CVCL_0125 | [4] |
| Half Maximal Inhibitory Concentration (IC50) | 0.02±0.001 µM | 72 h | N.A. | A2780 cell | CVCL_0134 | [5] |
| Half Maximal Inhibitory Concentration (IC50) | 0.028 µg/mL | 72 h | N.A. | MCF-7 cell | CVCL_0031 | [4] |
| Half Maximal Inhibitory Concentration (IC50) | 0.17±0.01 µM | 72 h | N.A. | PANC-1 cell | CVCL_0480 | [5] |
| 30% Inhibitory Concentration (IC30) | 3.6 uM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [6] |
| 30% Inhibitory Concentration (IC30) | 23 uM | N.A. | N.A. | DLD-1 cell | CVCL_0248 | [6] |
| 90% Growth Inhibition (GI90) | <4 ng/mL | N.A. | N.A. | A-549 cell | CVCL_0023 | [7] |
| 90% Growth Inhibition (GI90) | 5.81 ug/mL | N.A. | N.A. | THP-1 cell | CVCL_0006 | [8] |
| 90% Growth Inhibition (GI90) | 5.814 ug/mL | N.A. | N.A. | THP-1 cell | CVCL_0006 | [9] |
| 90% Growth Inhibition (GI90) | 38 ng/mL | N.A. | N.A. | A-549 cell | CVCL_0023 | [10] |
| 90% Growth Inhibition (GI90) | 70.6 ng/mL | N.A. | N.A. | A-549 cell | CVCL_0023 | [9] |
| 90% Lethal Concentration (IC50) | 0.9 ug/mL | N.A. | N.A. | PA-1 cell | CVCL_0479 | [11] |
| 90% Lethal Concentration (IC50) | 2.5 ug/mL | N.A. | N.A. | WRL68 cell | CVCL_0581 | [11] |
| 90% Lethal Concentration (IC50) | 10 ng/mL | N.A. | N.A. | COLO 320DM cell | CVCL_0219 | [11] |
| 90% Lethal Concentration (IC50) | 47 ng/mL | N.A. | N.A. | KB cell | CVCL_0372 | [11] |
| 90% Lethal Concentration (IC50) | 65 ng/mL | N.A. | N.A. | Caco-2 cell | CVCL_0025 | [11] |
| 90% Lethal Concentration (IC50) | 8 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [12] |
| 90% Lethal Concentration (IC50) | 204 nM | N.A. | N.A. | MKN45 cell | CVCL_0434 | [12] |
| 90% Lethal Concentration (IC50) | 229 nM | N.A. | N.A. | A498 cell | CVCL_1056 | [12] |
| 90% Lethal Concentration (IC50) | 372 nM | N.A. | N.A. | NCI-H630 cell | CVCL_1572 | [12] |
| 90% Lethal Concentration (IC50) | 832 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [12] |
| Half Maximal Cytotoxicity Concentration (CC50) | 10 ng/mL | N.A. | N.A. | HeLa cell | CVCL_0030 | [13] |
| Half Maximal Effective Concentration (EC50) | 0.21 ug/mL | N.A. | N.A. | HCT-8 cell | CVCL_2478 | [14] |
| Half Maximal Effective Concentration (EC50) | <5 ng/mL | N.A. | N.A. | DU145 cell | CVCL_0105 | [14] |
| Half Maximal Effective Concentration (EC50) | <5 ng/mL | N.A. | N.A. | A-549 cell | CVCL_0023 | [14] |
| Half Maximal Effective Concentration (EC50) | <5 ng/mL | N.A. | N.A. | KB cell | CVCL_0372 | [14] |
| Half Maximal Effective Concentration (EC50) | 7.2 ng/mL | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [14] |
| Half Maximal Effective Concentration (EC50) | 2 nM | N.A. | N.A. | KB cell | CVCL_0372 | [15] |
| Half Maximal Effective Concentration (EC50) | 18 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [16] |
| Half Maximal Effective Concentration (EC50) | 30 nM | N.A. | N.A. | T-47D cell | CVCL_0553 | [17] |
| Half Maximal Effective Concentration (EC50) | 35 nM | N.A. | N.A. | T-47D cell | CVCL_0553 | [18] |
| Half Maximal Effective Concentration (EC50) | 37 nM | N.A. | N.A. | T-47D cell | CVCL_0553 | [16] |
| Half Maximal Effective Concentration (EC50) | 54 nM | N.A. | N.A. | DLD-1 cell | CVCL_0248 | [19] |
| Half Maximal Effective Concentration (EC50) | 75 nM | N.A. | N.A. | DLD-1 cell | CVCL_0248 | [17] |
| Half Maximal Effective Concentration (EC50) | 98 nM | N.A. | N.A. | NCI-H1299 cell | CVCL_0060 | [19] |
| Half Maximal Effective Concentration (EC50) | 163 nM | N.A. | N.A. | NCI-H1299 cell | CVCL_0060 | [17] |
| Half Maximal Effective Dosage (ED50) | 1 ng/mL | N.A. | N.A. | KB cell | CVCL_0372 | [20] |
| Half Maximal Effective Dosage (ED50) | 2 ng/mL | N.A. | N.A. | A2780-1A9 cell | CVCL_H619 | [21] |
| Half Maximal Effective Dosage (ED50) | 2 ng/mL | N.A. | N.A. | KB cell | CVCL_0372 | [22] |
| Half Maximal Effective Dosage (ED50) | 5 ng/mL | N.A. | N.A. | A-549 cell | CVCL_0023 | [21] |
| Half Maximal Effective Dosage (ED50) | 10 pg/ml | N.A. | N.A. | KB cell | CVCL_0372 | [23] |
| Half Maximal Effective Dosage (ED50) | 11 ng/mL | N.A. | N.A. | HCT-8 cell | CVCL_2478 | [21] |
| Half Maximal Effective Dosage (ED50) | 41 ng/mL | N.A. | N.A. | 1A9/ptx-10 cell | CVCL_H620 | [20] |
| Half Maximal Effective Dosage (ED50) | 60 ng/mL | N.A. | N.A. | HCT-8 cell | CVCL_2478 | [22] |
| Half Maximal Effective Dosage (ED50) | 0.1 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [24] |
| Half Maximal Effective Dosage (ED50) | 1 nM | N.A. | N.A. | A2780-1A9 cell | CVCL_H619 | [25] |
| Half Maximal Effective Dosage (ED50) | 1 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [26] |
| Half Maximal Effective Dosage (ED50) | 1.3 nM | N.A. | N.A. | DU145 cell | CVCL_0105 | [25] |
| Half Maximal Effective Dosage (ED50) | 1.53 nM | N.A. | N.A. | NCI-ADR-RES cell | CVCL_1452 | [27] |
| Half Maximal Effective Dosage (ED50) | 2.3 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [25] |
| Half Maximal Effective Dosage (ED50) | 2.6 nM | N.A. | N.A. | LNCaP cell | CVCL_0395 | [25] |
| Half Maximal Effective Dosage (ED50) | 3 nM | N.A. | N.A. | DU145 cell | CVCL_0105 | [28] |
| Half Maximal Effective Dosage (ED50) | 3.23 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [27] |
| Half Maximal Effective Dosage (ED50) | 7 nM | N.A. | N.A. | KB cell | CVCL_0372 | [28] |
| Half Maximal Effective Dosage (ED50) | 8 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [28] |
| Half Maximal Effective Dosage (ED50) | 8.87 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [29] |
| Half Maximal Effective Dosage (ED50) | 15 nM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [30] |
| Half Maximal Effective Dosage (ED50) | 27 nM | N.A. | N.A. | B16 cell | CVCL_F936 | [31] |
| Half Maximal Effective Dosage (ED50) | 28 nM | N.A. | N.A. | B16 cell | CVCL_F936 | [32] |
| Half Maximal Effective Dosage (ED50) | 31.5 nM | N.A. | N.A. | B16 cell | CVCL_F936 | [33] |
| Half Maximal Effective Dosage (ED50) | 46 nM | N.A. | N.A. | Col2 cell | CVCL_D645 | [34] |
| Half Maximal Effective Dosage (ED50) | 90 nM | N.A. | N.A. | J774 cell | CVCL_4692 | [35] |
| Half Maximal Effective Dosage (ED50) | >100 nM | N.A. | N.A. | HCT-8 cell | CVCL_2478 | [29] |
| Half Maximal Effective Dosage (ED50) | >100 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [29] |
| Half Maximal Effective Dosage (ED50) | 0.54 uM | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [30] |
| Half Maximal Effective Dosage (ED50) | 0.7 uM | N.A. | N.A. | B16 cell | CVCL_F936 | [33] |
| Half Maximal Effective Dosage (ED50) | 2 uM | N.A. | N.A. | NCI-ADR-RES cell | CVCL_1452 | [36] |
| Half Maximal Effective Dosage (ED50) | >10 uM | N.A. | N.A. | LNCaP cell | CVCL_0395 | [37] |
| Half Maximal Effective Dosage (ED50) | >10 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [37] |
| Half Maximal Growth Inhibition (GI50) | <2.3 ng/mL | N.A. | N.A. | A-549 cell | CVCL_0023 | [38] |
| Half Maximal Growth Inhibition (GI50) | 2.93 ng/mL | N.A. | N.A. | P388 cell | CVCL_7222 | [39] |
| Half Maximal Growth Inhibition (GI50) | 3.5 ng/mL | N.A. | N.A. | A-549 cell | CVCL_0023 | [9] |
| Half Maximal Growth Inhibition (GI50) | 6.69 mM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [40] |
| Half Maximal Growth Inhibition (GI50) | 10 ng/mL | N.A. | N.A. | K562 cell | CVCL_0004 | [41] |
| Half Maximal Growth Inhibition (GI50) | <10 pM | N.A. | N.A. | A431 cell | CVCL_0037 | [42] |
| Half Maximal Growth Inhibition (GI50) | 15 pM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [43] |
| Half Maximal Growth Inhibition (GI50) | 17 pM | N.A. | N.A. | HBL-100 cell | CVCL_4362 | [44] |
| Half Maximal Growth Inhibition (GI50) | 33 pM | N.A. | N.A. | HeLa cell | CVCL_0030 | [44] |
| Half Maximal Growth Inhibition (GI50) | 0.27 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [44] |
| Half Maximal Growth Inhibition (GI50) | 0.71 nM | N.A. | N.A. | A2780-1A9 cell | CVCL_H619 | [45] |
| Half Maximal Growth Inhibition (GI50) | 1 nM | N.A. | N.A. | KB cell | CVCL_0372 | [46] |
| Half Maximal Growth Inhibition (GI50) | <1 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [47] |
| Half Maximal Growth Inhibition (GI50) | 1 nM | N.A. | N.A. | MDA-MB-435 cell | CVCL_0417 | [48] |
| Half Maximal Growth Inhibition (GI50) | 1.1 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [49] |
| Half Maximal Growth Inhibition (GI50) | 1.34 nM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [50] |
| Half Maximal Growth Inhibition (GI50) | 1.47 nM | N.A. | N.A. | DU145 cell | CVCL_0105 | [50] |
| Half Maximal Growth Inhibition (GI50) | 1.65 nM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [51] |
| Half Maximal Growth Inhibition (GI50) | 1.76 nM | N.A. | N.A. | COLO205 cell | CVCL_F402 | [52] |
| Half Maximal Growth Inhibition (GI50) | 1.76 nM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [50] |
| Half Maximal Growth Inhibition (GI50) | 1.9 nM | N.A. | N.A. | MDA-MB-435 cell | CVCL_0417 | [48] |
| Half Maximal Growth Inhibition (GI50) | 2 nM | N.A. | N.A. | RPMI-8226 cell | CVCL_7353 | [53] |
| Half Maximal Growth Inhibition (GI50) | 2.12 nM | N.A. | N.A. | U-937 cell | CVCL_0007 | [54] |
| Half Maximal Growth Inhibition (GI50) | 2.355 nM | N.A. | N.A. | A431 cell | CVCL_0037 | [55] |
| Half Maximal Growth Inhibition (GI50) | 2.512 nM | N.A. | N.A. | SR cell | CVCL_1711 | [56] |
| Half Maximal Growth Inhibition (GI50) | 2.512 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [56] |
| Half Maximal Growth Inhibition (GI50) | 2.512 nM | N.A. | N.A. | MDA-MB-435 cell | CVCL_0417 | [56] |
| Half Maximal Growth Inhibition (GI50) | 3 nM | N.A. | N.A. | 2008 cell | CVCL_0473 | [57] |
| Half Maximal Growth Inhibition (GI50) | 3 nM | N.A. | N.A. | COLO205 cell | CVCL_F402 | [58] |
| Half Maximal Growth Inhibition (GI50) | 3 nM | N.A. | N.A. | HCC 2998 cell | CVCL_1266 | [53] |
| Half Maximal Growth Inhibition (GI50) | 3 nM | N.A. | N.A. | U-251MG cell | CVCL_0021 | [59] |
| Half Maximal Growth Inhibition (GI50) | 3.162 nM | N.A. | N.A. | COLO205 cell | CVCL_F402 | [56] |
| Half Maximal Growth Inhibition (GI50) | 3.162 nM | N.A. | N.A. | Hs 578T cell | CVCL_0332 | [56] |
| Half Maximal Growth Inhibition (GI50) | 3.162 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [56] |
| Half Maximal Growth Inhibition (GI50) | 3.162 nM | N.A. | N.A. | SF539 cell | CVCL_1691 | [56] |
| Half Maximal Growth Inhibition (GI50) | 3.66 nM | N.A. | N.A. | SF539 cell | CVCL_1691 | [52] |
| Half Maximal Growth Inhibition (GI50) | 3.9 nM | N.A. | N.A. | NCI-H226 cell | CVCL_1544 | [60] |
| Half Maximal Growth Inhibition (GI50) | 3.981 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [56] |
| Half Maximal Growth Inhibition (GI50) | 3.981 nM | N.A. | N.A. | SK-MEL-5 cell | CVCL_0527 | [56] |
| Half Maximal Growth Inhibition (GI50) | 3.981 nM | N.A. | N.A. | HCC 2998 cell | CVCL_1266 | [56] |
| Half Maximal Growth Inhibition (GI50) | 3.981 nM | N.A. | N.A. | U-251MG cell | CVCL_0021 | [56] |
| Half Maximal Growth Inhibition (GI50) | 4 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [58] |
| Half Maximal Growth Inhibition (GI50) | 4 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [58] |
| Half Maximal Growth Inhibition (GI50) | 4 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [61] |
| Half Maximal Growth Inhibition (GI50) | 4 nM | N.A. | N.A. | SNB-75 cell | CVCL_1706 | [58] |
| Half Maximal Growth Inhibition (GI50) | 4 nM | N.A. | N.A. | KM12 cell | CVCL_1331 | [58] |
| Half Maximal Growth Inhibition (GI50) | 4 nM | N.A. | N.A. | SW620 cell | CVCL_0547 | [60] |
| Half Maximal Growth Inhibition (GI50) | 4 nM | N.A. | N.A. | MDA-MB-435 cell | CVCL_0417 | [61] |
| Half Maximal Growth Inhibition (GI50) | 5 nM | N.A. | N.A. | CCRF-CEM cell | CVCL_0207 | [61] |
| Half Maximal Growth Inhibition (GI50) | 5 nM | N.A. | N.A. | MOLT-4 cell | CVCL_0013 | [60] |
| Half Maximal Growth Inhibition (GI50) | 5 nM | N.A. | N.A. | LOX IMVI cell | CVCL_1381 | [61] |
| Half Maximal Growth Inhibition (GI50) | 5 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [59] |
| Half Maximal Growth Inhibition (GI50) | 5 nM | N.A. | N.A. | SF539 cell | CVCL_1691 | [61] |
| Half Maximal Growth Inhibition (GI50) | 5 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [60] |
| Half Maximal Growth Inhibition (GI50) | 5.012 nM | N.A. | N.A. | SK-MEL-2 cell | CVCL_0069 | [56] |
| Half Maximal Growth Inhibition (GI50) | 5.012 nM | N.A. | N.A. | OVCAR-8 cell | CVCL_1629 | [56] |
| Half Maximal Growth Inhibition (GI50) | 5.012 nM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [56] |
| Half Maximal Growth Inhibition (GI50) | 5.23 nM | N.A. | N.A. | DU145 cell | CVCL_0105 | [62] |
| Half Maximal Growth Inhibition (GI50) | 5.46 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [63] |
| Half Maximal Growth Inhibition (GI50) | 5.568 nM | N.A. | N.A. | DU145 cell | CVCL_0105 | [55] |
| Half Maximal Growth Inhibition (GI50) | 5.58 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [64] |
| Half Maximal Growth Inhibition (GI50) | 6.3 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [60] |
| Half Maximal Growth Inhibition (GI50) | 6.3 nM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [60] |
| Half Maximal Growth Inhibition (GI50) | 6.3 nM | N.A. | N.A. | NCI-H23 cell | CVCL_1547 | [60] |
| Half Maximal Growth Inhibition (GI50) | 6.3 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [60] |
| Half Maximal Growth Inhibition (GI50) | 6.3 nM | N.A. | N.A. | SR cell | CVCL_1711 | [61] |
| Half Maximal Growth Inhibition (GI50) | 6.3 nM | N.A. | N.A. | K562 cell | CVCL_0004 | [60] |
| Half Maximal Growth Inhibition (GI50) | 6.4 nM | N.A. | N.A. | KB cell | CVCL_0372 | [65] |
| Half Maximal Growth Inhibition (GI50) | 7 nM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [47] |
| Half Maximal Growth Inhibition (GI50) | 7 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [49] |
| Half Maximal Growth Inhibition (GI50) | 7.03 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [66] |
| Half Maximal Growth Inhibition (GI50) | 7.1 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [67] |
| Half Maximal Growth Inhibition (GI50) | 7.6 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [68] |
| Half Maximal Growth Inhibition (GI50) | 7.9 nM | N.A. | N.A. | COLO205 cell | CVCL_F402 | [60] |
| Half Maximal Growth Inhibition (GI50) | 7.9 nM | N.A. | N.A. | OVCAR-8 cell | CVCL_1629 | [61] |
| Half Maximal Growth Inhibition (GI50) | 7.943 nM | N.A. | N.A. | RXF 393 cell | CVCL_1673 | [56] |
| Half Maximal Growth Inhibition (GI50) | 8 nM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [58] |
| Half Maximal Growth Inhibition (GI50) | 8.18 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [69] |
| Half Maximal Growth Inhibition (GI50) | <10 nM | N.A. | N.A. | Jurkat cell | CVCL_0065 | [70] |
| Half Maximal Growth Inhibition (GI50) | 10 nM | N.A. | N.A. | B16 cell | CVCL_F936 | [71] |
| Half Maximal Growth Inhibition (GI50) | 10 nM | N.A. | N.A. | M14 cell | CVCL_1395 | [56] |
| Half Maximal Growth Inhibition (GI50) | <10 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [72] |
| Half Maximal Growth Inhibition (GI50) | 10 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [47] |
| Half Maximal Growth Inhibition (GI50) | <10 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [73] |
| Half Maximal Growth Inhibition (GI50) | 10 nM | N.A. | N.A. | SNU-398 cell | CVCL_0077 | [18] |
| Half Maximal Growth Inhibition (GI50) | 10 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [74] |
| Half Maximal Growth Inhibition (GI50) | <10 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [72] |
| Half Maximal Growth Inhibition (GI50) | 11 nM | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [42] |
| Half Maximal Growth Inhibition (GI50) | 12 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [75] |
| Half Maximal Growth Inhibition (GI50) | 12.5 nM | N.A. | N.A. | NCI-H322M cell | CVCL_1557 | [60] |
| Half Maximal Growth Inhibition (GI50) | 12.5 nM | N.A. | N.A. | T-47D cell | CVCL_0553 | [60] |
| Half Maximal Growth Inhibition (GI50) | 12.5 nM | N.A. | N.A. | MDA-MB-468 cell | CVCL_0419 | [61] |
| Half Maximal Growth Inhibition (GI50) | 12.5 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [60] |
| Half Maximal Growth Inhibition (GI50) | 13 nM | N.A. | N.A. | NCI-H322M cell | CVCL_1557 | [58] |
| Half Maximal Growth Inhibition (GI50) | 15 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [76] |
| Half Maximal Growth Inhibition (GI50) | 15.8 nM | N.A. | N.A. | RXF 393 cell | CVCL_1673 | [60] |
| Half Maximal Growth Inhibition (GI50) | <16 nM | N.A. | N.A. | DU145 cell | CVCL_0105 | [73] |
| Half Maximal Growth Inhibition (GI50) | <16 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [73] |
| Half Maximal Growth Inhibition (GI50) | <16 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [73] |
| Half Maximal Growth Inhibition (GI50) | 18.73 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [55] |
| Half Maximal Growth Inhibition (GI50) | 20 nM | N.A. | N.A. | OVCAR-3 cell | CVCL_0465 | [74] |
| Half Maximal Growth Inhibition (GI50) | 20 nM | N.A. | N.A. | B16-F1 cell | CVCL_0158 | [77] |
| Half Maximal Growth Inhibition (GI50) | 20 nM | N.A. | N.A. | NUGC-3 cell | CVCL_1612 | [78] |
| Half Maximal Growth Inhibition (GI50) | 20 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [71] |
| Half Maximal Growth Inhibition (GI50) | 24 nM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [75] |
| Half Maximal Growth Inhibition (GI50) | 25 nM | N.A. | N.A. | HOP-92 cell | CVCL_1286 | [61] |
| Half Maximal Growth Inhibition (GI50) | 25 nM | N.A. | N.A. | SF-295 cell | CVCL_1690 | [61] |
| Half Maximal Growth Inhibition (GI50) | 25 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [79] |
| Half Maximal Growth Inhibition (GI50) | 26 nM | N.A. | N.A. | T-47D cell | CVCL_0553 | [19] |
| Half Maximal Growth Inhibition (GI50) | 30 nM | N.A. | N.A. | Vero cell | CVCL_0059 | [59] |
| Half Maximal Growth Inhibition (GI50) | 30 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [78] |
| Half Maximal Growth Inhibition (GI50) | 30 nM | N.A. | N.A. | SiHa cell | CVCL_0032 | [71] |
| Half Maximal Growth Inhibition (GI50) | 31.62 nM | N.A. | N.A. | NCI-H226 cell | CVCL_1544 | [56] |
| Half Maximal Growth Inhibition (GI50) | 37 nM | N.A. | N.A. | M21 cell | CVCL_D031 | [76] |
| Half Maximal Growth Inhibition (GI50) | 40 nM | N.A. | N.A. | SNB-19 cell | CVCL_0535 | [74] |
| Half Maximal Growth Inhibition (GI50) | 50 nM | N.A. | N.A. | RXF 393 cell | CVCL_1673 | [74] |
| Half Maximal Growth Inhibition (GI50) | 50 nM | N.A. | N.A. | Malme-3M cell | CVCL_1438 | [60] |
| Half Maximal Growth Inhibition (GI50) | 51 nM | N.A. | N.A. | 1A9/ptx-22 cell | CVCL_H621 | [45] |
| Half Maximal Growth Inhibition (GI50) | 54 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [76] |
| Half Maximal Growth Inhibition (GI50) | 59 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [80] |
| Half Maximal Growth Inhibition (GI50) | 60 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [81] |
| Half Maximal Growth Inhibition (GI50) | 61 nM | N.A. | N.A. | SNU-398 cell | CVCL_0077 | [81] |
| Half Maximal Growth Inhibition (GI50) | 63 nM | N.A. | N.A. | MES-SA/Dx5 cell | CVCL_2598 | [82] |
| Half Maximal Growth Inhibition (GI50) | 79 nM | N.A. | N.A. | EKVX cell | CVCL_1195 | [61] |
| Half Maximal Growth Inhibition (GI50) | >100 nM | N.A. | N.A. | HCT-8 cell | CVCL_2478 | [62] |
| Half Maximal Growth Inhibition (GI50) | 125 nM | N.A. | N.A. | UO-31 cell | CVCL_1911 | [60] |
| Half Maximal Growth Inhibition (GI50) | 158 nM | N.A. | N.A. | Caki-1 cell | CVCL_0234 | [60] |
| Half Maximal Growth Inhibition (GI50) | 180 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [78] |
| Half Maximal Growth Inhibition (GI50) | 199.53 nM | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [56] |
| Half Maximal Growth Inhibition (GI50) | 206.7 nM | N.A. | N.A. | Huh-7 cell | CVCL_0336 | [55] |
| Half Maximal Growth Inhibition (GI50) | 300 nM | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [47] |
| Half Maximal Growth Inhibition (GI50) | 398 nM | N.A. | N.A. | ACHN cell | CVCL_1067 | [53] |
| Half Maximal Growth Inhibition (GI50) | 400 nM | N.A. | N.A. | SK-MEL-2 cell | CVCL_0069 | [60] |
| Half Maximal Growth Inhibition (GI50) | 400 nM | N.A. | N.A. | SK-MEL-28 cell | CVCL_0526 | [60] |
| Half Maximal Growth Inhibition (GI50) | 630 nM | N.A. | N.A. | OVCAR-4 cell | CVCL_1627 | [60] |
| Half Maximal Growth Inhibition (GI50) | 1000 nM | N.A. | N.A. | UO-31 cell | CVCL_1911 | [56] |
| Half Maximal Growth Inhibition (GI50) | 1.07 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [83] |
| Half Maximal Growth Inhibition (GI50) | 1.5 uM | N.A. | N.A. | SW1573 cell | CVCL_1720 | [84] |
| Half Maximal Growth Inhibition (GI50) | 1.6 uM | N.A. | N.A. | SW1573 cell | CVCL_1720 | [84] |
| Half Maximal Growth Inhibition (GI50) | 2.19 uM | N.A. | N.A. | SK-N-SH cell | CVCL_0531 | [83] |
| Half Maximal Growth Inhibition (GI50) | 2.82 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [83] |
| Half Maximal Growth Inhibition (GI50) | 3.16228 uM | N.A. | N.A. | NCI-ADR-RES cell | CVCL_1452 | [56] |
| Half Maximal Growth Inhibition (GI50) | 5.9 uM | N.A. | N.A. | Farage cell | CVCL_0214 | [75] |
| Half Maximal Growth Inhibition (GI50) | 5.943 uM | N.A. | N.A. | NCI-ADR-RES cell | CVCL_1452 | [85] |
| Half Maximal Growth Inhibition (GI50) | >15 uM | N.A. | N.A. | NCI-ADR-RES cell | CVCL_1452 | [86] |
| Half Maximal Growth Inhibition (GI50) | 20 uM | N.A. | N.A. | NCI-H226 cell | CVCL_1544 | [47] |
| Half Maximal Growth Inhibition (GI50) | >50 uM | N.A. | N.A. | NCI-ADR-RES cell | CVCL_1452 | [49] |
| Half Maximal Growth Inhibition (GI50) | 80 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [40] |
| Half Maximal Inhibitory Concentration (IC50) | <0.1 ug/mL | N.A. | N.A. | B16 cell | CVCL_F936 | [87] |
| Half Maximal Inhibitory Concentration (IC50) | 0.1 ng/mL | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [88] |
| Half Maximal Inhibitory Concentration (IC50) | 0.136 ug/mL | N.A. | N.A. | A2780 cell | CVCL_0134 | [89] |
| Half Maximal Inhibitory Concentration (IC50) | 0.18 ug/mL | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [90] |
| Half Maximal Inhibitory Concentration (IC50) | 0.2 ug/mL | N.A. | N.A. | P388 cell | CVCL_7222 | [91] |
| Half Maximal Inhibitory Concentration (IC50) | 0.27 ug/mL | N.A. | N.A. | P388 cell | CVCL_7222 | [92] |
| Half Maximal Inhibitory Concentration (IC50) | 0.29 ng/mL | N.A. | N.A. | A-549 cell | CVCL_0023 | [93] |
| Half Maximal Inhibitory Concentration (IC50) | 0.47 ug/mL | N.A. | N.A. | HeLa cell | CVCL_0030 | [94] |
| Half Maximal Inhibitory Concentration (IC50) | 0.61 ug/mL | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [94] |
| Half Maximal Inhibitory Concentration (IC50) | 0.61 ug/mL | N.A. | N.A. | HeLa cell | CVCL_0030 | [95] |
| Half Maximal Inhibitory Concentration (IC50) | 0.69 ug/mL | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [96] |
| Half Maximal Inhibitory Concentration (IC50) | 1 ng/mL | N.A. | N.A. | CCRF-CEM cell | CVCL_0207 | [97] |
| Half Maximal Inhibitory Concentration (IC50) | 1.01 mM | N.A. | N.A. | MV4-11 cell | CVCL_0064 | [98] |
| Half Maximal Inhibitory Concentration (IC50) | 1.18 ug/mL | N.A. | N.A. | HeLa cell | CVCL_0030 | [99] |
| Half Maximal Inhibitory Concentration (IC50) | 1.3 ug/mL | N.A. | N.A. | SiHa cell | CVCL_0032 | [87] |
| Half Maximal Inhibitory Concentration (IC50) | 1.34 ug/mL | N.A. | N.A. | EL4 cell | CVCL_0255 | [88] |
| Half Maximal Inhibitory Concentration (IC50) | 1.6 ng/mL | N.A. | N.A. | KB cell | CVCL_0372 | [100] |
| Half Maximal Inhibitory Concentration (IC50) | 1.9 ng/mL | N.A. | N.A. | A-549 cell | CVCL_0023 | [100] |
| Half Maximal Inhibitory Concentration (IC50) | 2 ng/mL | N.A. | N.A. | DU145 cell | CVCL_0105 | [101] |
| Half Maximal Inhibitory Concentration (IC50) | 2 ug/mL | N.A. | N.A. | CCD 19Lu cell | CVCL_2382 | [97] |
| Half Maximal Inhibitory Concentration (IC50) | <2 ug/mL | N.A. | N.A. | A-549 cell | CVCL_0023 | [102] |
| Half Maximal Inhibitory Concentration (IC50) | 2 ng/mL | N.A. | N.A. | A-549 cell | CVCL_0023 | [101] |
| Half Maximal Inhibitory Concentration (IC50) | 2 ng/mL | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [91] |
| Half Maximal Inhibitory Concentration (IC50) | 2.4 ng/mL | N.A. | N.A. | DU145 cell | CVCL_0105 | [100] |
| Half Maximal Inhibitory Concentration (IC50) | 2.4 ng/mL | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [103] |
| Half Maximal Inhibitory Concentration (IC50) | <3 ng/mL | N.A. | N.A. | A498 cell | CVCL_1056 | [104] |
| Half Maximal Inhibitory Concentration (IC50) | <3 ng/mL | N.A. | N.A. | NCI-H226 cell | CVCL_1544 | [104] |
| Half Maximal Inhibitory Concentration (IC50) | <3 ng/mL | N.A. | N.A. | M19-MEL cell | CVCL_B415 | [104] |
| Half Maximal Inhibitory Concentration (IC50) | <3 ng/mL | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [104] |
| Half Maximal Inhibitory Concentration (IC50) | <3 ng/mL | N.A. | N.A. | WiDr cell | CVCL_2760 | [104] |
| Half Maximal Inhibitory Concentration (IC50) | 3.4 ng/mL | N.A. | N.A. | A-549 cell | CVCL_0023 | [103] |
| Half Maximal Inhibitory Concentration (IC50) | 3.5 ng/mL | N.A. | N.A. | WRL68 cell | CVCL_0581 | [11] |
| Half Maximal Inhibitory Concentration (IC50) | 3.9 ug/mL | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [105] |
| Half Maximal Inhibitory Concentration (IC50) | 4 ug/mL | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [105] |
| Half Maximal Inhibitory Concentration (IC50) | 4.1 ng/mL | N.A. | N.A. | B16-F10 cell | CVCL_0159 | [103] |
| Half Maximal Inhibitory Concentration (IC50) | 4.3 ng/mL | N.A. | N.A. | WI-38 VA13 cell | CVCL_2759 | [106] |
| Half Maximal Inhibitory Concentration (IC50) | 4.5 ng/mL | N.A. | N.A. | COLO 320DM cell | CVCL_0219 | [11] |
| Half Maximal Inhibitory Concentration (IC50) | 4.8 ng/mL | N.A. | N.A. | KB cell | CVCL_0372 | [107] |
| Half Maximal Inhibitory Concentration (IC50) | 5.3 ng/mL | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [108] |
| Half Maximal Inhibitory Concentration (IC50) | 6.9 ug/mL | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [106] |
| Half Maximal Inhibitory Concentration (IC50) | 7.8 ng/mL | N.A. | N.A. | Caco-2 cell | CVCL_0025 | [109] |
| Half Maximal Inhibitory Concentration (IC50) | 8 ng/mL | N.A. | N.A. | HEK293 cell | CVCL_0045 | [99] |
| Half Maximal Inhibitory Concentration (IC50) | 8 ng/mL | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [109] |
| Half Maximal Inhibitory Concentration (IC50) | 8 ng/mL | N.A. | N.A. | HeLa cell | CVCL_0030 | [110] |
| Half Maximal Inhibitory Concentration (IC50) | 8 ng/mL | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [111] |
| Half Maximal Inhibitory Concentration (IC50) | 8.1 mM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [112] |
| Half Maximal Inhibitory Concentration (IC50) | 8.7 ug/mL | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [87] |
| Half Maximal Inhibitory Concentration (IC50) | 10 ng/mL | N.A. | N.A. | IGROV-1 cell | CVCL_1304 | [104] |
| Half Maximal Inhibitory Concentration (IC50) | 10 ng/mL | N.A. | N.A. | P388 cell | CVCL_7222 | [97] |
| Half Maximal Inhibitory Concentration (IC50) | 10 ng/mL | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [88] |
| Half Maximal Inhibitory Concentration (IC50) | 10.6 pg/ml | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [113] |
| Half Maximal Inhibitory Concentration (IC50) | 19.64 ng/mL | N.A. | N.A. | A-549 cell | CVCL_0023 | [114] |
| Half Maximal Inhibitory Concentration (IC50) | 20 ng/mL | N.A. | N.A. | WI-38 cell | CVCL_0579 | [115] |
| Half Maximal Inhibitory Concentration (IC50) | <20 ng/mL | N.A. | N.A. | B16 cell | CVCL_F936 | [116] |
| Half Maximal Inhibitory Concentration (IC50) | <20 ng/mL | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [116] |
| Half Maximal Inhibitory Concentration (IC50) | 30 ng/mL | N.A. | N.A. | P388 cell | CVCL_7222 | [117] |
| Half Maximal Inhibitory Concentration (IC50) | 30 ng/mL | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [118] |
| Half Maximal Inhibitory Concentration (IC50) | 46 ng/mL | N.A. | N.A. | P388 cell | CVCL_7222 | [103] |
| Half Maximal Inhibitory Concentration (IC50) | 50 ng/mL | N.A. | N.A. | P388 cell | CVCL_7222 | [119] |
| Half Maximal Inhibitory Concentration (IC50) | 50 ng/mL | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [115] |
| Half Maximal Inhibitory Concentration (IC50) | >85 ng/mL | N.A. | N.A. | KB cell | CVCL_0372 | [101] |
| Half Maximal Inhibitory Concentration (IC50) | >400 ug/mL | N.A. | N.A. | HeLa cell | CVCL_0030 | [120] |
| Half Maximal Inhibitory Concentration (IC50) | 0.11 pM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [121] |
| Half Maximal Inhibitory Concentration (IC50) | 40 pM | N.A. | N.A. | KB cell | CVCL_0372 | [122] |
| Half Maximal Inhibitory Concentration (IC50) | 90 pM | N.A. | N.A. | L1210 cell | CVCL_0382 | [123] |
| Half Maximal Inhibitory Concentration (IC50) | 0.2 nM | N.A. | N.A. | ARO cell | CVCL_0144 | [124] |
| Half Maximal Inhibitory Concentration (IC50) | 0.2 nM | N.A. | N.A. | MDA-MB-435 cell | CVCL_0417 | [125] |
| Half Maximal Inhibitory Concentration (IC50) | 0.27 nM | N.A. | N.A. | CCRF-CEM cell | CVCL_0207 | [126] |
| Half Maximal Inhibitory Concentration (IC50) | 0.32 nM | N.A. | N.A. | WM 266-4 cell | CVCL_2765 | [121] |
| Half Maximal Inhibitory Concentration (IC50) | 0.4 nM | N.A. | N.A. | L1210 cell | CVCL_0382 | [126] |
| Half Maximal Inhibitory Concentration (IC50) | 0.43 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [127] |
| Half Maximal Inhibitory Concentration (IC50) | 0.44 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [128] |
| Half Maximal Inhibitory Concentration (IC50) | 0.5 nM | N.A. | N.A. | Jurkat cell | CVCL_0065 | [129] |
| Half Maximal Inhibitory Concentration (IC50) | 0.5 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [123] |
| Half Maximal Inhibitory Concentration (IC50) | 0.5 nM | N.A. | N.A. | MDA-MB-435 cell | CVCL_0417 | [130] |
| Half Maximal Inhibitory Concentration (IC50) | 0.52 nM | N.A. | N.A. | MRC5 cell | CVCL_0440 | [131] |
| Half Maximal Inhibitory Concentration (IC50) | 0.58 nM | N.A. | N.A. | L1210 cell | CVCL_0382 | [123] |
| Half Maximal Inhibitory Concentration (IC50) | 0.6 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [132] |
| Half Maximal Inhibitory Concentration (IC50) | 0.6 nM | N.A. | N.A. | SNU-398 cell | CVCL_0077 | [133] |
| Half Maximal Inhibitory Concentration (IC50) | 0.6 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [134] |
| Half Maximal Inhibitory Concentration (IC50) | 0.62 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [135] |
| Half Maximal Inhibitory Concentration (IC50) | 0.7 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [106] |
| Half Maximal Inhibitory Concentration (IC50) | 0.71 nM | N.A. | N.A. | K562 cell | CVCL_0004 | [136] |
| Half Maximal Inhibitory Concentration (IC50) | 0.8 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [137] |
| Half Maximal Inhibitory Concentration (IC50) | 0.82 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [138] |
| Half Maximal Inhibitory Concentration (IC50) | 0.91 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [139] |
| Half Maximal Inhibitory Concentration (IC50) | <1 nM | N.A. | N.A. | BGC-823 cell | CVCL_3360 | [140] |
| Half Maximal Inhibitory Concentration (IC50) | 1 nM | N.A. | N.A. | BGC-823 cell | CVCL_3360 | [141] |
| Half Maximal Inhibitory Concentration (IC50) | <1 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [140] |
| Half Maximal Inhibitory Concentration (IC50) | 1 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [142] |
| Half Maximal Inhibitory Concentration (IC50) | 1 nM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [143] |
| Half Maximal Inhibitory Concentration (IC50) | 1 nM | N.A. | N.A. | KB cell | CVCL_0372 | [144] |
| Half Maximal Inhibitory Concentration (IC50) | 1 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [145] |
| Half Maximal Inhibitory Concentration (IC50) | 1 nM | N.A. | N.A. | SW48 cell | CVCL_1724 | [146] |
| Half Maximal Inhibitory Concentration (IC50) | 1.07 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [147] |
| Half Maximal Inhibitory Concentration (IC50) | 1.1 nM | N.A. | N.A. | CCRF-CEM cell | CVCL_0207 | [148] |
| Half Maximal Inhibitory Concentration (IC50) | 1.1 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [147] |
| Half Maximal Inhibitory Concentration (IC50) | 1.1 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [149] |
| Half Maximal Inhibitory Concentration (IC50) | 1.1 nM | N.A. | N.A. | SW780 cell | CVCL_1728 | [150] |
| Half Maximal Inhibitory Concentration (IC50) | 1.25 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [151] |
| Half Maximal Inhibitory Concentration (IC50) | 1.274 nM | N.A. | N.A. | CCRF-CEM cell | CVCL_0207 | [152] |
| Half Maximal Inhibitory Concentration (IC50) | 1.3 nM | N.A. | N.A. | BGC-823 cell | CVCL_3360 | [153] |
| Half Maximal Inhibitory Concentration (IC50) | 1.3 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [154] |
| Half Maximal Inhibitory Concentration (IC50) | 1.3 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [155] |
| Half Maximal Inhibitory Concentration (IC50) | 1.38 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [156] |
| Half Maximal Inhibitory Concentration (IC50) | 1.4 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [157] |
| Half Maximal Inhibitory Concentration (IC50) | 1.4 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [158] |
| Half Maximal Inhibitory Concentration (IC50) | 1.4 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [159] |
| Half Maximal Inhibitory Concentration (IC50) | 1.5 nM | N.A. | N.A. | CCRF-CEM cell | CVCL_0207 | [160] |
| Half Maximal Inhibitory Concentration (IC50) | 1.5 nM | N.A. | N.A. | OVCAR-3 cell | CVCL_0465 | [137] |
| Half Maximal Inhibitory Concentration (IC50) | 1.5 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [161] |
| Half Maximal Inhibitory Concentration (IC50) | 1.5 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [162] |
| Half Maximal Inhibitory Concentration (IC50) | 1.6 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [163] |
| Half Maximal Inhibitory Concentration (IC50) | 1.6 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [164] |
| Half Maximal Inhibitory Concentration (IC50) | 1.7 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [165] |
| Half Maximal Inhibitory Concentration (IC50) | 1.7 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [166] |
| Half Maximal Inhibitory Concentration (IC50) | 1.8 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [167] |
| Half Maximal Inhibitory Concentration (IC50) | 1.9 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [12] |
| Half Maximal Inhibitory Concentration (IC50) | 1.9 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [168] |
| Half Maximal Inhibitory Concentration (IC50) | 1.93 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [169] |
| Half Maximal Inhibitory Concentration (IC50) | 1.98 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [170] |
| Half Maximal Inhibitory Concentration (IC50) | 2 nM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [171] |
| Half Maximal Inhibitory Concentration (IC50) | 2 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [172] |
| Half Maximal Inhibitory Concentration (IC50) | 2 nM | N.A. | N.A. | OVCAR-8 cell | CVCL_1629 | [173] |
| Half Maximal Inhibitory Concentration (IC50) | 2 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [174] |
| Half Maximal Inhibitory Concentration (IC50) | 2 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [175] |
| Half Maximal Inhibitory Concentration (IC50) | 2 nM | N.A. | N.A. | SW480 cell | CVCL_0546 | [146] |
| Half Maximal Inhibitory Concentration (IC50) | 2 nM | N.A. | N.A. | L02 cell | CVCL_6926 | [176] |
| Half Maximal Inhibitory Concentration (IC50) | 2.1 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [162] |
| Half Maximal Inhibitory Concentration (IC50) | 2.12 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [177] |
| Half Maximal Inhibitory Concentration (IC50) | 2.2 nM | N.A. | N.A. | MOLT-4 cell | CVCL_0013 | [178] |
| Half Maximal Inhibitory Concentration (IC50) | 2.4 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [12] |
| Half Maximal Inhibitory Concentration (IC50) | 2.45 nM | N.A. | N.A. | KB cell | CVCL_0372 | [179] |
| Half Maximal Inhibitory Concentration (IC50) | 2.5 nM | N.A. | N.A. | H69AR cell | CVCL_3513 | [180] |
| Half Maximal Inhibitory Concentration (IC50) | 2.5 nM | N.A. | N.A. | NCI-H1975 cell | CVCL_1511 | [181] |
| Half Maximal Inhibitory Concentration (IC50) | 2.5 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [182] |
| Half Maximal Inhibitory Concentration (IC50) | 2.5 nM | N.A. | N.A. | KB cell | CVCL_0372 | [183] |
| Half Maximal Inhibitory Concentration (IC50) | 2.6 nM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [136] |
| Half Maximal Inhibitory Concentration (IC50) | 2.6 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [184] |
| Half Maximal Inhibitory Concentration (IC50) | 2.6 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [185] |
| Half Maximal Inhibitory Concentration (IC50) | 2.7 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [186] |
| Half Maximal Inhibitory Concentration (IC50) | 2.8 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [187] |
| Half Maximal Inhibitory Concentration (IC50) | 2.8 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [188] |
| Half Maximal Inhibitory Concentration (IC50) | 2.92 nM | N.A. | N.A. | KB 3-1 cell | CVCL_2088 | [189] |
| Half Maximal Inhibitory Concentration (IC50) | 2.95 nM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [190] |
| Half Maximal Inhibitory Concentration (IC50) | 3 nM | N.A. | N.A. | CCRF-CEM cell | CVCL_0207 | [155] |
| Half Maximal Inhibitory Concentration (IC50) | 3 nM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [61] |
| Half Maximal Inhibitory Concentration (IC50) | 3 nM | N.A. | N.A. | PA-1 cell | CVCL_0479 | [191] |
| Half Maximal Inhibitory Concentration (IC50) | 3 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [192] |
| Half Maximal Inhibitory Concentration (IC50) | 3 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [193] |
| Half Maximal Inhibitory Concentration (IC50) | 3 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [194] |
| Half Maximal Inhibitory Concentration (IC50) | 3 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [184] |
| Half Maximal Inhibitory Concentration (IC50) | 3 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [195] |
| Half Maximal Inhibitory Concentration (IC50) | 3.01 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [177] |
| Half Maximal Inhibitory Concentration (IC50) | 3.1 nM | N.A. | N.A. | CCRF-CEM cell | CVCL_0207 | [196] |
| Half Maximal Inhibitory Concentration (IC50) | 3.1 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [151] |
| Half Maximal Inhibitory Concentration (IC50) | 3.1 nM | N.A. | N.A. | MDA-MB-435 cell | CVCL_0417 | [197] |
| Half Maximal Inhibitory Concentration (IC50) | 3.14 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [139] |
| Half Maximal Inhibitory Concentration (IC50) | 3.2 nM | N.A. | N.A. | SJSA-1 cell | CVCL_1697 | [198] |
| Half Maximal Inhibitory Concentration (IC50) | 3.2 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [165] |
| Half Maximal Inhibitory Concentration (IC50) | 3.21 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [159] |
| Half Maximal Inhibitory Concentration (IC50) | 3.29 nM | N.A. | N.A. | BGC-823 cell | CVCL_3360 | [199] |
| Half Maximal Inhibitory Concentration (IC50) | 3.3 nM | N.A. | N.A. | BGC-823 cell | CVCL_3360 | [200] |
| Half Maximal Inhibitory Concentration (IC50) | 3.3 nM | N.A. | N.A. | OVCAR-3 cell | CVCL_0465 | [201] |
| Half Maximal Inhibitory Concentration (IC50) | 3.3 nM | N.A. | N.A. | COLO205 cell | CVCL_F402 | [202] |
| Half Maximal Inhibitory Concentration (IC50) | 3.3 nM | N.A. | N.A. | KB cell | CVCL_0372 | [203] |
| Half Maximal Inhibitory Concentration (IC50) | 3.3 nM | N.A. | N.A. | MDA-MB-435 cell | CVCL_0417 | [204] |
| Half Maximal Inhibitory Concentration (IC50) | 3.388 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [205] |
| Half Maximal Inhibitory Concentration (IC50) | 3.4 nM | N.A. | N.A. | DU145 cell | CVCL_0105 | [178] |
| Half Maximal Inhibitory Concentration (IC50) | 3.4 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [206] |
| Half Maximal Inhibitory Concentration (IC50) | 3.4 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [186] |
| Half Maximal Inhibitory Concentration (IC50) | 3.4 nM | N.A. | N.A. | U2OS cell | CVCL_0042 | [207] |
| Half Maximal Inhibitory Concentration (IC50) | 3.4 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [208] |
| Half Maximal Inhibitory Concentration (IC50) | 3.4 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [209] |
| Half Maximal Inhibitory Concentration (IC50) | 3.5 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [98] |
| Half Maximal Inhibitory Concentration (IC50) | 3.6 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [165] |
| Half Maximal Inhibitory Concentration (IC50) | 3.6 nM | N.A. | N.A. | HCT-8 cell | CVCL_2478 | [200] |
| Half Maximal Inhibitory Concentration (IC50) | 3.6 nM | N.A. | N.A. | KB cell | CVCL_0372 | [210] |
| Half Maximal Inhibitory Concentration (IC50) | 3.6 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [186] |
| Half Maximal Inhibitory Concentration (IC50) | 3.7 nM | N.A. | N.A. | OVCAR-8 cell | CVCL_1629 | [181] |
| Half Maximal Inhibitory Concentration (IC50) | 3.8 nM | N.A. | N.A. | MGC-803 cell | CVCL_5334 | [153] |
| Half Maximal Inhibitory Concentration (IC50) | 3.8 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [208] |
| Half Maximal Inhibitory Concentration (IC50) | 3.9 nM | N.A. | N.A. | KB 3-1 cell | CVCL_2088 | [211] |
| Half Maximal Inhibitory Concentration (IC50) | 3.99 nM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [212] |
| Half Maximal Inhibitory Concentration (IC50) | 4 nM | N.A. | N.A. | CCRF-CEM cell | CVCL_0207 | [213] |
| Half Maximal Inhibitory Concentration (IC50) | 4 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [214] |
| Half Maximal Inhibitory Concentration (IC50) | 4 nM | N.A. | N.A. | T-47D cell | CVCL_0553 | [146] |
| Half Maximal Inhibitory Concentration (IC50) | 4 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [215] |
| Half Maximal Inhibitory Concentration (IC50) | 4 nM | N.A. | N.A. | OVCAR-8 cell | CVCL_1629 | [216] |
| Half Maximal Inhibitory Concentration (IC50) | 4 nM | N.A. | N.A. | WRL68 cell | CVCL_0581 | [217] |
| Half Maximal Inhibitory Concentration (IC50) | 4 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [218] |
| Half Maximal Inhibitory Concentration (IC50) | 4 nM | N.A. | N.A. | MDA-MB-435 cell | CVCL_0417 | [219] |
| Half Maximal Inhibitory Concentration (IC50) | 4.1 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [186] |
| Half Maximal Inhibitory Concentration (IC50) | 4.1 nM | N.A. | N.A. | KB cell | CVCL_0372 | [220] |
| Half Maximal Inhibitory Concentration (IC50) | 4.2 nM | N.A. | N.A. | KB cell | CVCL_0372 | [221] |
| Half Maximal Inhibitory Concentration (IC50) | 4.3 nM | N.A. | N.A. | WI-38 VA13 cell | CVCL_2759 | [222] |
| Half Maximal Inhibitory Concentration (IC50) | 4.4 nM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [163] |
| Half Maximal Inhibitory Concentration (IC50) | 4.4 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [223] |
| Half Maximal Inhibitory Concentration (IC50) | 4.4 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [224] |
| Half Maximal Inhibitory Concentration (IC50) | 4.5 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [118] |
| Half Maximal Inhibitory Concentration (IC50) | 4.6 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [225] |
| Half Maximal Inhibitory Concentration (IC50) | 4.6 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [226] |
| Half Maximal Inhibitory Concentration (IC50) | 4.6 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [227] |
| Half Maximal Inhibitory Concentration (IC50) | 4.6 nM | N.A. | N.A. | A-375 cell | CVCL_0132 | [223] |
| Half Maximal Inhibitory Concentration (IC50) | 4.7 nM | N.A. | N.A. | OVCAR-8 cell | CVCL_1629 | [228] |
| Half Maximal Inhibitory Concentration (IC50) | 4.8 nM | N.A. | N.A. | A2780-1A9 cell | CVCL_H619 | [229] |
| Half Maximal Inhibitory Concentration (IC50) | 4.89 nM | N.A. | N.A. | DU145 cell | CVCL_0105 | [230] |
| Half Maximal Inhibitory Concentration (IC50) | 4.9 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [12] |
| Half Maximal Inhibitory Concentration (IC50) | 4.9 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [231] |
| Half Maximal Inhibitory Concentration (IC50) | 4.9 nM | N.A. | N.A. | A-375 cell | CVCL_0132 | [199] |
| Half Maximal Inhibitory Concentration (IC50) | 5 nM | N.A. | N.A. | MES-SA cell | CVCL_1404 | [232] |
| Half Maximal Inhibitory Concentration (IC50) | 5 nM | N.A. | N.A. | DU145 cell | CVCL_0105 | [233] |
| Half Maximal Inhibitory Concentration (IC50) | 5 nM | N.A. | N.A. | PT-45 cell | CVCL_8407 | [124] |
| Half Maximal Inhibitory Concentration (IC50) | 5 nM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [12] |
| Half Maximal Inhibitory Concentration (IC50) | 5 nM | N.A. | N.A. | WI-38 VA13 cell | CVCL_2759 | [234] |
| Half Maximal Inhibitory Concentration (IC50) | 5 nM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [235] |
| Half Maximal Inhibitory Concentration (IC50) | 5 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [175] |
| Half Maximal Inhibitory Concentration (IC50) | 5 nM | N.A. | N.A. | MDA-MB-468 cell | CVCL_0419 | [181] |
| Half Maximal Inhibitory Concentration (IC50) | 5 nM | N.A. | N.A. | MDA-MB-435 cell | CVCL_0417 | [232] |
| Half Maximal Inhibitory Concentration (IC50) | 5 nM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [233] |
| Half Maximal Inhibitory Concentration (IC50) | 5.3 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [225] |
| Half Maximal Inhibitory Concentration (IC50) | 5.3 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [236] |
| Half Maximal Inhibitory Concentration (IC50) | 5.5 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [237] |
| Half Maximal Inhibitory Concentration (IC50) | 5.6 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [238] |
| Half Maximal Inhibitory Concentration (IC50) | 5.7 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [239] |
| Half Maximal Inhibitory Concentration (IC50) | 5.71 nM | N.A. | N.A. | KB cell | CVCL_0372 | [240] |
| Half Maximal Inhibitory Concentration (IC50) | 5.9 nM | N.A. | N.A. | WI-38 VA13 cell | CVCL_2759 | [241] |
| Half Maximal Inhibitory Concentration (IC50) | 5.9 nM | N.A. | N.A. | U-937/GTB cell | CVCL_U631 | [242] |
| Half Maximal Inhibitory Concentration (IC50) | 6 nM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [243] |
| Half Maximal Inhibitory Concentration (IC50) | 6 nM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [244] |
| Half Maximal Inhibitory Concentration (IC50) | 6 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [245] |
| Half Maximal Inhibitory Concentration (IC50) | 6 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [246] |
| Half Maximal Inhibitory Concentration (IC50) | 6 nM | N.A. | N.A. | KB cell | CVCL_0372 | [247] |
| Half Maximal Inhibitory Concentration (IC50) | 6 nM | N.A. | N.A. | Bel-7402 cell | CVCL_5492 | [140] |
| Half Maximal Inhibitory Concentration (IC50) | 6 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [195] |
| Half Maximal Inhibitory Concentration (IC50) | 6 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [248] |
| Half Maximal Inhibitory Concentration (IC50) | 6 nM | N.A. | N.A. | KB 3-1 cell | CVCL_2088 | [249] |
| Half Maximal Inhibitory Concentration (IC50) | 6 nM | N.A. | N.A. | BT-549 cell | CVCL_1092 | [243] |
| Half Maximal Inhibitory Concentration (IC50) | 6.1 nM | N.A. | N.A. | A121 cell | CVCL_G294 | [165] |
| Half Maximal Inhibitory Concentration (IC50) | 6.1 nM | N.A. | N.A. | MDA-MB-468 cell | CVCL_0419 | [250] |
| Half Maximal Inhibitory Concentration (IC50) | 6.2 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [251] |
| Half Maximal Inhibitory Concentration (IC50) | 6.3 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [252] |
| Half Maximal Inhibitory Concentration (IC50) | 6.3 nM | N.A. | N.A. | A121 cell | CVCL_G294 | [186] |
| Half Maximal Inhibitory Concentration (IC50) | 6.3 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [253] |
| Half Maximal Inhibitory Concentration (IC50) | 6.3 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [254] |
| Half Maximal Inhibitory Concentration (IC50) | 6.6 nM | N.A. | N.A. | KB cell | CVCL_0372 | [255] |
| Half Maximal Inhibitory Concentration (IC50) | 6.6 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [256] |
| Half Maximal Inhibitory Concentration (IC50) | 6.8 nM | N.A. | N.A. | NCI-H226 cell | CVCL_1544 | [257] |
| Half Maximal Inhibitory Concentration (IC50) | 6.8 nM | N.A. | N.A. | KB cell | CVCL_0372 | [258] |
| Half Maximal Inhibitory Concentration (IC50) | 7 nM | N.A. | N.A. | BGC-823 cell | CVCL_3360 | [252] |
| Half Maximal Inhibitory Concentration (IC50) | 7 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [259] |
| Half Maximal Inhibitory Concentration (IC50) | 7 nM | N.A. | N.A. | PANC-1 cell | CVCL_0480 | [260] |
| Half Maximal Inhibitory Concentration (IC50) | 7 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [261] |
| Half Maximal Inhibitory Concentration (IC50) | 7 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [124] |
| Half Maximal Inhibitory Concentration (IC50) | 7 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [262] |
| Half Maximal Inhibitory Concentration (IC50) | 7 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [263] |
| Half Maximal Inhibitory Concentration (IC50) | 7.2 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [264] |
| Half Maximal Inhibitory Concentration (IC50) | 7.2 nM | N.A. | N.A. | SK-MEL-2 cell | CVCL_0069 | [12] |
| Half Maximal Inhibitory Concentration (IC50) | 7.2 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [265] |
| Half Maximal Inhibitory Concentration (IC50) | 7.3 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [266] |
| Half Maximal Inhibitory Concentration (IC50) | 7.32 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [177] |
| Half Maximal Inhibitory Concentration (IC50) | 7.5 nM | N.A. | N.A. | A2780S cell | CVCL_4863 | [267] |
| Half Maximal Inhibitory Concentration (IC50) | 7.52 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [240] |
| Half Maximal Inhibitory Concentration (IC50) | 7.53 nM | N.A. | N.A. | NCI-N87 cell | CVCL_1603 | [268] |
| Half Maximal Inhibitory Concentration (IC50) | 7.7 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [269] |
| Half Maximal Inhibitory Concentration (IC50) | 7.9 nM | N.A. | N.A. | YOSHIDA cell | CVCL_G359 | [231] |
| Half Maximal Inhibitory Concentration (IC50) | 7.9 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [200] |
| Half Maximal Inhibitory Concentration (IC50) | 7.9 nM | N.A. | N.A. | GES1 cell | CVCL_EQ22 | [153] |
| Half Maximal Inhibitory Concentration (IC50) | 7.94 nM | N.A. | N.A. | NCI-H1975 cell | CVCL_1511 | [268] |
| Half Maximal Inhibitory Concentration (IC50) | <8 nM | N.A. | N.A. | PANC-1 cell | CVCL_0480 | [270] |
| Half Maximal Inhibitory Concentration (IC50) | <8 nM | N.A. | N.A. | NCM460 cell | CVCL_0460 | [271] |
| Half Maximal Inhibitory Concentration (IC50) | <8 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [272] |
| Half Maximal Inhibitory Concentration (IC50) | <8 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [273] |
| Half Maximal Inhibitory Concentration (IC50) | <8 nM | N.A. | N.A. | SMMC-7721 cell | CVCL_0534 | [274] |
| Half Maximal Inhibitory Concentration (IC50) | 8 nM | N.A. | N.A. | Huh-7 cell | CVCL_0336 | [275] |
| Half Maximal Inhibitory Concentration (IC50) | 8 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [276] |
| Half Maximal Inhibitory Concentration (IC50) | 8 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [277] |
| Half Maximal Inhibitory Concentration (IC50) | <8 nM | N.A. | N.A. | SW480 cell | CVCL_0546 | [271] |
| Half Maximal Inhibitory Concentration (IC50) | <8 nM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [278] |
| Half Maximal Inhibitory Concentration (IC50) | 8.2 nM | N.A. | N.A. | NCI-H520 cell | CVCL_1566 | [191] |
| Half Maximal Inhibitory Concentration (IC50) | 8.2 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [254] |
| Half Maximal Inhibitory Concentration (IC50) | 8.37 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [240] |
| Half Maximal Inhibitory Concentration (IC50) | 8.4 nM | N.A. | N.A. | HGC-27 cell | CVCL_1279 | [153] |
| Half Maximal Inhibitory Concentration (IC50) | 8.51 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [240] |
| Half Maximal Inhibitory Concentration (IC50) | 8.54 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [240] |
| Half Maximal Inhibitory Concentration (IC50) | 8.76 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [240] |
| Half Maximal Inhibitory Concentration (IC50) | 8.8 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [279] |
| Half Maximal Inhibitory Concentration (IC50) | 8.8 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [251] |
| Half Maximal Inhibitory Concentration (IC50) | 8.9 nM | N.A. | N.A. | A-375 cell | CVCL_0132 | [280] |
| Half Maximal Inhibitory Concentration (IC50) | 9 nM | N.A. | N.A. | EMT6 cell | CVCL_1923 | [281] |
| Half Maximal Inhibitory Concentration (IC50) | 9 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [192] |
| Half Maximal Inhibitory Concentration (IC50) | 9.1 nM | N.A. | N.A. | KB cell | CVCL_0372 | [237] |
| Half Maximal Inhibitory Concentration (IC50) | 9.1 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [282] |
| Half Maximal Inhibitory Concentration (IC50) | 9.1 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [283] |
| Half Maximal Inhibitory Concentration (IC50) | 9.2 nM | N.A. | N.A. | A431 cell | CVCL_0037 | [15] |
| Half Maximal Inhibitory Concentration (IC50) | 9.5 nM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [284] |
| Half Maximal Inhibitory Concentration (IC50) | 9.9 nM | N.A. | N.A. | PANC-1 cell | CVCL_0480 | [285] |
| Half Maximal Inhibitory Concentration (IC50) | 10 nM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [286] |
| Half Maximal Inhibitory Concentration (IC50) | 10 nM | N.A. | N.A. | OVCAR-3 cell | CVCL_0465 | [124] |
| Half Maximal Inhibitory Concentration (IC50) | 10 nM | N.A. | N.A. | P388 cell | CVCL_7222 | [233] |
| Half Maximal Inhibitory Concentration (IC50) | >10 nM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [287] |
| Half Maximal Inhibitory Concentration (IC50) | 10 nM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [288] |
| Half Maximal Inhibitory Concentration (IC50) | >10 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [240] |
| Half Maximal Inhibitory Concentration (IC50) | 10 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [240] |
| Half Maximal Inhibitory Concentration (IC50) | 10 nM | N.A. | N.A. | KB cell | CVCL_0372 | [289] |
| Half Maximal Inhibitory Concentration (IC50) | 10 nM | N.A. | N.A. | OVCAR-8 cell | CVCL_1629 | [290] |
| Half Maximal Inhibitory Concentration (IC50) | 10 nM | N.A. | N.A. | RKO cell | CVCL_0504 | [286] |
| Half Maximal Inhibitory Concentration (IC50) | 10 nM | N.A. | N.A. | SF268 cell | CVCL_1689 | [291] |
| Half Maximal Inhibitory Concentration (IC50) | 10 nM | N.A. | N.A. | A-375 cell | CVCL_0132 | [288] |
| Half Maximal Inhibitory Concentration (IC50) | 10 nM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [292] |
| Half Maximal Inhibitory Concentration (IC50) | 11 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [293] |
| Half Maximal Inhibitory Concentration (IC50) | 11 nM | N.A. | N.A. | SW480 cell | CVCL_0546 | [243] |
| Half Maximal Inhibitory Concentration (IC50) | 11.2 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [258] |
| Half Maximal Inhibitory Concentration (IC50) | 11.4 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [255] |
| Half Maximal Inhibitory Concentration (IC50) | 11.46 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [294] |
| Half Maximal Inhibitory Concentration (IC50) | 12 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [295] |
| Half Maximal Inhibitory Concentration (IC50) | 12 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [215] |
| Half Maximal Inhibitory Concentration (IC50) | 12 nM | N.A. | N.A. | T98G cell | CVCL_0556 | [296] |
| Half Maximal Inhibitory Concentration (IC50) | 12.06 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [297] |
| Half Maximal Inhibitory Concentration (IC50) | 12.1 nM | N.A. | N.A. | SW480 cell | CVCL_0546 | [254] |
| Half Maximal Inhibitory Concentration (IC50) | 12.1 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [240] |
| Half Maximal Inhibitory Concentration (IC50) | 12.3 nM | N.A. | N.A. | A2780S cell | CVCL_4863 | [298] |
| Half Maximal Inhibitory Concentration (IC50) | 13 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [299] |
| Half Maximal Inhibitory Concentration (IC50) | 13.4 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [227] |
| Half Maximal Inhibitory Concentration (IC50) | 13.7 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [300] |
| Half Maximal Inhibitory Concentration (IC50) | 13.7 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [168] |
| Half Maximal Inhibitory Concentration (IC50) | 14 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [301] |
| Half Maximal Inhibitory Concentration (IC50) | 14 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [280] |
| Half Maximal Inhibitory Concentration (IC50) | 14.9 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [157] |
| Half Maximal Inhibitory Concentration (IC50) | 15 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [229] |
| Half Maximal Inhibitory Concentration (IC50) | 15 nM | N.A. | N.A. | OV-1063 cell | CVCL_4366 | [281] |
| Half Maximal Inhibitory Concentration (IC50) | 15 nM | N.A. | N.A. | NCI-H1299 cell | CVCL_0060 | [118] |
| Half Maximal Inhibitory Concentration (IC50) | 15.2 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [302] |
| Half Maximal Inhibitory Concentration (IC50) | 15.3 nM | N.A. | N.A. | SF268 cell | CVCL_1689 | [303] |
| Half Maximal Inhibitory Concentration (IC50) | 16 nM | N.A. | N.A. | P388 cell | CVCL_7222 | [304] |
| Half Maximal Inhibitory Concentration (IC50) | 16 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [305] |
| Half Maximal Inhibitory Concentration (IC50) | 16 nM | N.A. | N.A. | MDA-MB-435 cell | CVCL_0417 | [127] |
| Half Maximal Inhibitory Concentration (IC50) | 17 nM | N.A. | N.A. | A498 cell | CVCL_1056 | [12] |
| Half Maximal Inhibitory Concentration (IC50) | 17 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [306] |
| Half Maximal Inhibitory Concentration (IC50) | 17 nM | N.A. | N.A. | MV4-11 cell | CVCL_0064 | [307] |
| Half Maximal Inhibitory Concentration (IC50) | 17.3 nM | N.A. | N.A. | LOX IMVI cell | CVCL_1381 | [211] |
| Half Maximal Inhibitory Concentration (IC50) | 17.4 nM | N.A. | N.A. | HCT-8 cell | CVCL_2478 | [298] |
| Half Maximal Inhibitory Concentration (IC50) | 18 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [122] |
| Half Maximal Inhibitory Concentration (IC50) | 18.9 nM | N.A. | N.A. | BGC-823 cell | CVCL_3360 | [308] |
| Half Maximal Inhibitory Concentration (IC50) | 19 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [252] |
| Half Maximal Inhibitory Concentration (IC50) | 19 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [309] |
| Half Maximal Inhibitory Concentration (IC50) | 19.7 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [310] |
| Half Maximal Inhibitory Concentration (IC50) | 20 nM | N.A. | N.A. | BGC-823 cell | CVCL_3360 | [311] |
| Half Maximal Inhibitory Concentration (IC50) | 20 nM | N.A. | N.A. | P388 cell | CVCL_7222 | [312] |
| Half Maximal Inhibitory Concentration (IC50) | 20 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [312] |
| Half Maximal Inhibitory Concentration (IC50) | 20 nM | N.A. | N.A. | KB cell | CVCL_0372 | [312] |
| Half Maximal Inhibitory Concentration (IC50) | 20 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [313] |
| Half Maximal Inhibitory Concentration (IC50) | 20 nM | N.A. | N.A. | RKO cell | CVCL_0504 | [314] |
| Half Maximal Inhibitory Concentration (IC50) | 20.5 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [223] |
| Half Maximal Inhibitory Concentration (IC50) | 21 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [315] |
| Half Maximal Inhibitory Concentration (IC50) | 21 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [214] |
| Half Maximal Inhibitory Concentration (IC50) | 21.1 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [316] |
| Half Maximal Inhibitory Concentration (IC50) | 21.7 nM | N.A. | N.A. | SF268 cell | CVCL_1689 | [317] |
| Half Maximal Inhibitory Concentration (IC50) | 22 nM | N.A. | N.A. | DLD-1 cell | CVCL_0248 | [285] |
| Half Maximal Inhibitory Concentration (IC50) | 23 nM | N.A. | N.A. | IGROV-1 cell | CVCL_1304 | [207] |
| Half Maximal Inhibitory Concentration (IC50) | 23 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [318] |
| Half Maximal Inhibitory Concentration (IC50) | 23.1 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [303] |
| Half Maximal Inhibitory Concentration (IC50) | 23.18 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [319] |
| Half Maximal Inhibitory Concentration (IC50) | 23.4 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [320] |
| Half Maximal Inhibitory Concentration (IC50) | 24 nM | N.A. | N.A. | U2OS cell | CVCL_0042 | [161] |
| Half Maximal Inhibitory Concentration (IC50) | 24 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [321] |
| Half Maximal Inhibitory Concentration (IC50) | 25 nM | N.A. | N.A. | ACHN cell | CVCL_1067 | [180] |
| Half Maximal Inhibitory Concentration (IC50) | 25 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [118] |
| Half Maximal Inhibitory Concentration (IC50) | 25.1 nM | N.A. | N.A. | A431 cell | CVCL_0037 | [322] |
| Half Maximal Inhibitory Concentration (IC50) | 26 nM | N.A. | N.A. | NCI-H838 cell | CVCL_1594 | [323] |
| Half Maximal Inhibitory Concentration (IC50) | 27 nM | N.A. | N.A. | DLD-1 cell | CVCL_0248 | [304] |
| Half Maximal Inhibitory Concentration (IC50) | 28 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [324] |
| Half Maximal Inhibitory Concentration (IC50) | 28 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [325] |
| Half Maximal Inhibitory Concentration (IC50) | 28 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [318] |
| Half Maximal Inhibitory Concentration (IC50) | 30 nM | N.A. | N.A. | NB-4 cell | CVCL_0005 | [326] |
| Half Maximal Inhibitory Concentration (IC50) | 30 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [141] |
| Half Maximal Inhibitory Concentration (IC50) | 30 nM | N.A. | N.A. | U2OS cell | CVCL_0042 | [327] |
| Half Maximal Inhibitory Concentration (IC50) | 30 nM | N.A. | N.A. | P388/ADR cell | CVCL_IZ75 | [328] |
| Half Maximal Inhibitory Concentration (IC50) | 30 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [329] |
| Half Maximal Inhibitory Concentration (IC50) | 33.8 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [330] |
| Half Maximal Inhibitory Concentration (IC50) | 35 nM | N.A. | N.A. | NCI-H596 cell | CVCL_1571 | [176] |
| Half Maximal Inhibitory Concentration (IC50) | 35 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [331] |
| Half Maximal Inhibitory Concentration (IC50) | 35 nM | N.A. | N.A. | MX1 cell | CVCL_4774 | [155] |
| Half Maximal Inhibitory Concentration (IC50) | 37 nM | N.A. | N.A. | HCT-8 cell | CVCL_2478 | [252] |
| Half Maximal Inhibitory Concentration (IC50) | 37.7 nM | N.A. | N.A. | NCI-H1299 cell | CVCL_0060 | [211] |
| Half Maximal Inhibitory Concentration (IC50) | 40 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [98] |
| Half Maximal Inhibitory Concentration (IC50) | 40 nM | N.A. | N.A. | WI-38 cell | CVCL_0579 | [332] |
| Half Maximal Inhibitory Concentration (IC50) | 40 nM | N.A. | N.A. | P388 cell | CVCL_7222 | [333] |
| Half Maximal Inhibitory Concentration (IC50) | 40 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [334] |
| Half Maximal Inhibitory Concentration (IC50) | 43 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [325] |
| Half Maximal Inhibitory Concentration (IC50) | 46 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [307] |
| Half Maximal Inhibitory Concentration (IC50) | 46 nM | N.A. | N.A. | MX1 cell | CVCL_4774 | [149] |
| Half Maximal Inhibitory Concentration (IC50) | 46.8 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [335] |
| Half Maximal Inhibitory Concentration (IC50) | 47 nM | N.A. | N.A. | CFPAC-1 cell | CVCL_1119 | [336] |
| Half Maximal Inhibitory Concentration (IC50) | 50 nM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [314] |
| Half Maximal Inhibitory Concentration (IC50) | 50 nM | N.A. | N.A. | P388 cell | CVCL_7222 | [314] |
| Half Maximal Inhibitory Concentration (IC50) | 50 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [337] |
| Half Maximal Inhibitory Concentration (IC50) | 50 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [124] |
| Half Maximal Inhibitory Concentration (IC50) | 50 nM | N.A. | N.A. | DLD-1 cell | CVCL_0248 | [243] |
| Half Maximal Inhibitory Concentration (IC50) | 50 nM | N.A. | N.A. | SF268 cell | CVCL_1689 | [314] |
| Half Maximal Inhibitory Concentration (IC50) | 51 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [307] |
| Half Maximal Inhibitory Concentration (IC50) | 51 nM | N.A. | N.A. | HCT-8 cell | CVCL_2478 | [140] |
| Half Maximal Inhibitory Concentration (IC50) | 52 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [338] |
| Half Maximal Inhibitory Concentration (IC50) | 53 nM | N.A. | N.A. | AGS cell | CVCL_0139 | [153] |
| Half Maximal Inhibitory Concentration (IC50) | 58 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [339] |
| Half Maximal Inhibitory Concentration (IC50) | 60 nM | N.A. | N.A. | IGROV-1 cell | CVCL_1304 | [340] |
| Half Maximal Inhibitory Concentration (IC50) | 60 nM | N.A. | N.A. | L1210 cell | CVCL_0382 | [333] |
| Half Maximal Inhibitory Concentration (IC50) | 60 nM | N.A. | N.A. | Bel-7402 cell | CVCL_5492 | [341] |
| Half Maximal Inhibitory Concentration (IC50) | 61 nM | N.A. | N.A. | A498 cell | CVCL_1056 | [257] |
| Half Maximal Inhibitory Concentration (IC50) | 62.01 nM | N.A. | N.A. | OVCAR-3 cell | CVCL_0465 | [342] |
| Half Maximal Inhibitory Concentration (IC50) | 63.3 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [239] |
| Half Maximal Inhibitory Concentration (IC50) | 65.6 nM | N.A. | N.A. | J774 cell | CVCL_4692 | [343] |
| Half Maximal Inhibitory Concentration (IC50) | 65.8 nM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [322] |
| Half Maximal Inhibitory Concentration (IC50) | 66 nM | N.A. | N.A. | ACHN cell | CVCL_1067 | [288] |
| Half Maximal Inhibitory Concentration (IC50) | 70 nM | N.A. | N.A. | P388 cell | CVCL_7222 | [314] |
| Half Maximal Inhibitory Concentration (IC50) | 70 nM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [314] |
| Half Maximal Inhibitory Concentration (IC50) | 70.3 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [335] |
| Half Maximal Inhibitory Concentration (IC50) | 77 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [118] |
| Half Maximal Inhibitory Concentration (IC50) | 80 nM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [344] |
| Half Maximal Inhibitory Concentration (IC50) | 80 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [345] |
| Half Maximal Inhibitory Concentration (IC50) | 80 nM | N.A. | N.A. | KB cell | CVCL_0372 | [346] |
| Half Maximal Inhibitory Concentration (IC50) | <80 nM | N.A. | N.A. | OS-RC-2 cell | CVCL_1626 | [347] |
| Half Maximal Inhibitory Concentration (IC50) | 82 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [348] |
| Half Maximal Inhibitory Concentration (IC50) | 84 nM | N.A. | N.A. | H22 cell | CVCL_H613 | [349] |
| Half Maximal Inhibitory Concentration (IC50) | 90 nM | N.A. | N.A. | DLD-1 cell | CVCL_0248 | [350] |
| Half Maximal Inhibitory Concentration (IC50) | 100 nM | N.A. | N.A. | NB-4 cell | CVCL_0005 | [351] |
| Half Maximal Inhibitory Concentration (IC50) | 100 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [352] |
| Half Maximal Inhibitory Concentration (IC50) | 100 nM | N.A. | N.A. | Bel-7402 cell | CVCL_5492 | [252] |
| Half Maximal Inhibitory Concentration (IC50) | 100 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [175] |
| Half Maximal Inhibitory Concentration (IC50) | 105 nM | N.A. | N.A. | MDA-MB-435 cell | CVCL_0417 | [328] |
| Half Maximal Inhibitory Concentration (IC50) | 110 nM | N.A. | N.A. | HEp-2 cell | CVCL_1906 | [353] |
| Half Maximal Inhibitory Concentration (IC50) | 110 nM | N.A. | N.A. | SK-BR-3 cell | CVCL_0033 | [354] |
| Half Maximal Inhibitory Concentration (IC50) | 120 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [353] |
| Half Maximal Inhibitory Concentration (IC50) | 120 nM | N.A. | N.A. | L1210 cell | CVCL_0382 | [355] |
| Half Maximal Inhibitory Concentration (IC50) | 128 nM | N.A. | N.A. | U-251MG cell | CVCL_0021 | [356] |
| Half Maximal Inhibitory Concentration (IC50) | 140 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [357] |
| Half Maximal Inhibitory Concentration (IC50) | 140 nM | N.A. | N.A. | CHO-VV 3-2 cell | CVCL_Y287 | [136] |
| Half Maximal Inhibitory Concentration (IC50) | 157 nM | N.A. | N.A. | 1A9/ptx-10 cell | CVCL_H620 | [229] |
| Half Maximal Inhibitory Concentration (IC50) | 170 nM | N.A. | N.A. | CHO cell | CVCL_0213 | [126] |
| Half Maximal Inhibitory Concentration (IC50) | 170 nM | N.A. | N.A. | HCT-8 cell | CVCL_2478 | [344] |
| Half Maximal Inhibitory Concentration (IC50) | 180 nM | N.A. | N.A. | SW1990 cell | CVCL_1723 | [235] |
| Half Maximal Inhibitory Concentration (IC50) | 180 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [358] |
| Half Maximal Inhibitory Concentration (IC50) | 188 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [127] |
| Half Maximal Inhibitory Concentration (IC50) | 190 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [329] |
| Half Maximal Inhibitory Concentration (IC50) | 200 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [326] |
| Half Maximal Inhibitory Concentration (IC50) | 200 nM | N.A. | N.A. | 4T1 cell | CVCL_0125 | [245] |
| Half Maximal Inhibitory Concentration (IC50) | 200 nM | N.A. | N.A. | SH-SY5Y cell | CVCL_0019 | [359] |
| Half Maximal Inhibitory Concentration (IC50) | 200 nM | N.A. | N.A. | OE33 cell | CVCL_0471 | [124] |
| Half Maximal Inhibitory Concentration (IC50) | 240 nM | N.A. | N.A. | HCT-8 cell | CVCL_2478 | [235] |
| Half Maximal Inhibitory Concentration (IC50) | 240 nM | N.A. | N.A. | SK-HEP1 cell | CVCL_0525 | [357] |
| Half Maximal Inhibitory Concentration (IC50) | 273 nM | N.A. | N.A. | KB cell | CVCL_0372 | [360] |
| Half Maximal Inhibitory Concentration (IC50) | 290 nM | N.A. | N.A. | SF-295 cell | CVCL_1690 | [303] |
| Half Maximal Inhibitory Concentration (IC50) | 299 nM | N.A. | N.A. | NCI-ADR-RES cell | CVCL_1452 | [361] |
| Half Maximal Inhibitory Concentration (IC50) | 299 nM | N.A. | N.A. | MCF7R cell | CVCL_Y493 | [362] |
| Half Maximal Inhibitory Concentration (IC50) | 300 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [165] |
| Half Maximal Inhibitory Concentration (IC50) | 300 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [124] |
| Half Maximal Inhibitory Concentration (IC50) | 310 nM | N.A. | N.A. | MDA-MB-468 cell | CVCL_0419 | [363] |
| Half Maximal Inhibitory Concentration (IC50) | 312 nM | N.A. | N.A. | SW480 cell | CVCL_0546 | [364] |
| Half Maximal Inhibitory Concentration (IC50) | 350 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [365] |
| Half Maximal Inhibitory Concentration (IC50) | 379 nM | N.A. | N.A. | MDA-MB-435 cell | CVCL_0417 | [219] |
| Half Maximal Inhibitory Concentration (IC50) | 380 nM | N.A. | N.A. | OVCAR-3 cell | CVCL_0465 | [366] |
| Half Maximal Inhibitory Concentration (IC50) | 380 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [347] |
| Half Maximal Inhibitory Concentration (IC50) | 380 nM | N.A. | N.A. | K562 cell | CVCL_0004 | [367] |
| Half Maximal Inhibitory Concentration (IC50) | 390 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [358] |
| Half Maximal Inhibitory Concentration (IC50) | 400 nM | N.A. | N.A. | CCRF-CEM cell | CVCL_0207 | [149] |
| Half Maximal Inhibitory Concentration (IC50) | 400 nM | N.A. | N.A. | OE19 cell | CVCL_1622 | [124] |
| Half Maximal Inhibitory Concentration (IC50) | 410 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [366] |
| Half Maximal Inhibitory Concentration (IC50) | 410 nM | N.A. | N.A. | K562 cell | CVCL_0004 | [368] |
| Half Maximal Inhibitory Concentration (IC50) | 429 nM | N.A. | N.A. | CCRF-CEM cell | CVCL_0207 | [196] |
| Half Maximal Inhibitory Concentration (IC50) | 430 nM | N.A. | N.A. | CCRF-CEM cell | CVCL_0207 | [155] |
| Half Maximal Inhibitory Concentration (IC50) | 438 nM | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [211] |
| Half Maximal Inhibitory Concentration (IC50) | 450 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [357] |
| Half Maximal Inhibitory Concentration (IC50) | 470 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [350] |
| Half Maximal Inhibitory Concentration (IC50) | 484.3 nM | N.A. | N.A. | CCRF-CEM cell | CVCL_0207 | [162] |
| Half Maximal Inhibitory Concentration (IC50) | 490 nM | N.A. | N.A. | MDA-MB-435 cell | CVCL_0417 | [369] |
| Half Maximal Inhibitory Concentration (IC50) | 500 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [370] |
| Half Maximal Inhibitory Concentration (IC50) | 520 nM | N.A. | N.A. | CHO-TAX 5-6 cell | CVCL_U346 | [126] |
| Half Maximal Inhibitory Concentration (IC50) | 547 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [186] |
| Half Maximal Inhibitory Concentration (IC50) | 550 nM | N.A. | N.A. | MCF7R cell | CVCL_Y493 | [371] |
| Half Maximal Inhibitory Concentration (IC50) | 578.2 nM | N.A. | N.A. | Bel-7402 cell | CVCL_5492 | [223] |
| Half Maximal Inhibitory Concentration (IC50) | 590 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [372] |
| Half Maximal Inhibitory Concentration (IC50) | 600 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [373] |
| Half Maximal Inhibitory Concentration (IC50) | 612 nM | N.A. | N.A. | MDA-MB-435 cell | CVCL_0417 | [369] |
| Half Maximal Inhibitory Concentration (IC50) | 620 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [374] |
| Half Maximal Inhibitory Concentration (IC50) | 650 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [375] |
| Half Maximal Inhibitory Concentration (IC50) | 650 nM | N.A. | N.A. | Vero cell | CVCL_0059 | [129] |
| Half Maximal Inhibitory Concentration (IC50) | 660 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [357] |
| Half Maximal Inhibitory Concentration (IC50) | 670 nM | N.A. | N.A. | HCT-8 cell | CVCL_2478 | [375] |
| Half Maximal Inhibitory Concentration (IC50) | 680 nM | N.A. | N.A. | A-375 cell | CVCL_0132 | [376] |
| Half Maximal Inhibitory Concentration (IC50) | 690 nM | N.A. | N.A. | Jurkat cell | CVCL_0065 | [363] |
| Half Maximal Inhibitory Concentration (IC50) | 700 nM | N.A. | N.A. | UACC-812 cell | CVCL_1781 | [146] |
| Half Maximal Inhibitory Concentration (IC50) | 740 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [363] |
| Half Maximal Inhibitory Concentration (IC50) | 759.39 nM | N.A. | N.A. | A549/TR cell | CVCL_C5S0 | [319] |
| Half Maximal Inhibitory Concentration (IC50) | 810 nM | N.A. | N.A. | A-375 cell | CVCL_0132 | [346] |
| Half Maximal Inhibitory Concentration (IC50) | 820 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [377] |
| Half Maximal Inhibitory Concentration (IC50) | 830 nM | N.A. | N.A. | MGC-803 cell | CVCL_5334 | [367] |
| Half Maximal Inhibitory Concentration (IC50) | 850 nM | N.A. | N.A. | MGC-803 cell | CVCL_5334 | [378] |
| Half Maximal Inhibitory Concentration (IC50) | 860 nM | N.A. | N.A. | MCF7R cell | CVCL_Y493 | [379] |
| Half Maximal Inhibitory Concentration (IC50) | 890 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [378] |
| Half Maximal Inhibitory Concentration (IC50) | 900 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [380] |
| Half Maximal Inhibitory Concentration (IC50) | 930 nM | N.A. | N.A. | Bel-7402 cell | CVCL_5492 | [381] |
| Half Maximal Inhibitory Concentration (IC50) | >1000 nM | N.A. | N.A. | NCI-H1650 cell | CVCL_1483 | [382] |
| Half Maximal Inhibitory Concentration (IC50) | 1.03 uM | N.A. | N.A. | MEXF514 cell | CVCL_D129 | [334] |
| Half Maximal Inhibitory Concentration (IC50) | 1.04 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [383] |
| Half Maximal Inhibitory Concentration (IC50) | 1.16 uM | N.A. | N.A. | K562 cell | CVCL_0004 | [384] |
| Half Maximal Inhibitory Concentration (IC50) | 1.17 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [385] |
| Half Maximal Inhibitory Concentration (IC50) | 1.2 uM | N.A. | N.A. | CCRF-CEM cell | CVCL_0207 | [149] |
| Half Maximal Inhibitory Concentration (IC50) | 1.239 uM | N.A. | N.A. | A2780 cell | CVCL_0134 | [264] |
| Half Maximal Inhibitory Concentration (IC50) | 1.25 uM | N.A. | N.A. | Bel-7402 cell | CVCL_5492 | [337] |
| Half Maximal Inhibitory Concentration (IC50) | 1.27 uM | N.A. | N.A. | CCRF-CEM cell | CVCL_0207 | [386] |
| Half Maximal Inhibitory Concentration (IC50) | 1.3 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [346] |
| Half Maximal Inhibitory Concentration (IC50) | 1.35 uM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [387] |
| Half Maximal Inhibitory Concentration (IC50) | 1.375 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [254] |
| Half Maximal Inhibitory Concentration (IC50) | 1.428 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [388] |
| Half Maximal Inhibitory Concentration (IC50) | 1.5 uM | N.A. | N.A. | BGC-823 cell | CVCL_3360 | [347] |
| Half Maximal Inhibitory Concentration (IC50) | 1.5 uM | N.A. | N.A. | NCI-H1299 cell | CVCL_0060 | [125] |
| Half Maximal Inhibitory Concentration (IC50) | 1.51 uM | N.A. | N.A. | A549/TR cell | CVCL_C5S0 | [389] |
| Half Maximal Inhibitory Concentration (IC50) | 1.7 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [390] |
| Half Maximal Inhibitory Concentration (IC50) | 1.8 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [391] |
| Half Maximal Inhibitory Concentration (IC50) | 1.8 uM | N.A. | N.A. | Bel-7402 cell | CVCL_5492 | [375] |
| Half Maximal Inhibitory Concentration (IC50) | 1.89 uM | N.A. | N.A. | Bel-7402 cell | CVCL_5492 | [392] |
| Half Maximal Inhibitory Concentration (IC50) | >2 uM | N.A. | N.A. | Bel-7402 cell | CVCL_5492 | [357] |
| Half Maximal Inhibitory Concentration (IC50) | 2.1 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [393] |
| Half Maximal Inhibitory Concentration (IC50) | 2.11 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [394] |
| Half Maximal Inhibitory Concentration (IC50) | 2.29087 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [330] |
| Half Maximal Inhibitory Concentration (IC50) | 2.4 uM | N.A. | N.A. | K562 cell | CVCL_0004 | [395] |
| Half Maximal Inhibitory Concentration (IC50) | 2.43 uM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [396] |
| Half Maximal Inhibitory Concentration (IC50) | 2.46 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [384] |
| Half Maximal Inhibitory Concentration (IC50) | 2.5 uM | N.A. | N.A. | MIA PaCa-2 cell | CVCL_0428 | [376] |
| Half Maximal Inhibitory Concentration (IC50) | 2.56 uM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [397] |
| Half Maximal Inhibitory Concentration (IC50) | 2.6 uM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [398] |
| Half Maximal Inhibitory Concentration (IC50) | 2.655 uM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [399] |
| Half Maximal Inhibitory Concentration (IC50) | 2.7 uM | N.A. | N.A. | MES-SA/Dx5 cell | CVCL_2598 | [400] |
| Half Maximal Inhibitory Concentration (IC50) | 2.7 uM | N.A. | N.A. | HEY cell | CVCL_0297 | [212] |
| Half Maximal Inhibitory Concentration (IC50) | 2.7 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [401] |
| Half Maximal Inhibitory Concentration (IC50) | 2.7 uM | N.A. | N.A. | K562 cell | CVCL_0004 | [402] |
| Half Maximal Inhibitory Concentration (IC50) | 2.85 uM | N.A. | N.A. | A2780 cell | CVCL_0134 | [385] |
| Half Maximal Inhibitory Concentration (IC50) | 2.9 uM | N.A. | N.A. | Ca-Ski cell | CVCL_1100 | [403] |
| Half Maximal Inhibitory Concentration (IC50) | 2.98 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [396] |
| Half Maximal Inhibitory Concentration (IC50) | >3 uM | N.A. | N.A. | RKO cell | CVCL_0504 | [286] |
| Half Maximal Inhibitory Concentration (IC50) | >3 uM | N.A. | N.A. | KB 3-1 cell | CVCL_2088 | [249] |
| Half Maximal Inhibitory Concentration (IC50) | 3.164 uM | N.A. | N.A. | A2780/Taxol cell | CVCL_IJ13 | [267] |
| Half Maximal Inhibitory Concentration (IC50) | 3.2 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [395] |
| Half Maximal Inhibitory Concentration (IC50) | 3.29 uM | N.A. | N.A. | HCT-8 cell | CVCL_2478 | [396] |
| Half Maximal Inhibitory Concentration (IC50) | 3.6 uM | N.A. | N.A. | HCT-8 cell | CVCL_2478 | [404] |
| Half Maximal Inhibitory Concentration (IC50) | 3.8 uM | N.A. | N.A. | KB cell | CVCL_0372 | [405] |
| Half Maximal Inhibitory Concentration (IC50) | 3.8 uM | N.A. | N.A. | K562 cell | CVCL_0004 | [393] |
| Half Maximal Inhibitory Concentration (IC50) | 3.8 uM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [393] |
| Half Maximal Inhibitory Concentration (IC50) | 3.87 uM | N.A. | N.A. | MDA-MB-468 cell | CVCL_0419 | [397] |
| Half Maximal Inhibitory Concentration (IC50) | 4.14 uM | N.A. | N.A. | K562 cell | CVCL_0004 | [406] |
| Half Maximal Inhibitory Concentration (IC50) | 4.15 uM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [406] |
| Half Maximal Inhibitory Concentration (IC50) | 4.37 uM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [384] |
| Half Maximal Inhibitory Concentration (IC50) | 4.57 uM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [407] |
| Half Maximal Inhibitory Concentration (IC50) | 4.73 uM | N.A. | N.A. | SK-MEL-24 cell | CVCL_0599 | [408] |
| Half Maximal Inhibitory Concentration (IC50) | 4.73 uM | N.A. | N.A. | SK-MEL3 cell | CVCL_0550 | [409] |
| Half Maximal Inhibitory Concentration (IC50) | 4.77 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [378] |
| Half Maximal Inhibitory Concentration (IC50) | 4.84 uM | N.A. | N.A. | DU145 cell | CVCL_0105 | [410] |
| Half Maximal Inhibitory Concentration (IC50) | 4.875 uM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [190] |
| Half Maximal Inhibitory Concentration (IC50) | 4.875 uM | N.A. | N.A. | NIH3T3 cell | CVCL_0594 | [411] |
| Half Maximal Inhibitory Concentration (IC50) | 4.9 uM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [181] |
| Half Maximal Inhibitory Concentration (IC50) | 5 uM | N.A. | N.A. | MES-SA/Dx5 cell | CVCL_2598 | [232] |
| Half Maximal Inhibitory Concentration (IC50) | 5 uM | N.A. | N.A. | WI-38 VA13 cell | CVCL_2759 | [112] |
| Half Maximal Inhibitory Concentration (IC50) | >5 uM | N.A. | N.A. | P388/ADR cell | CVCL_IZ75 | [289] |
| Half Maximal Inhibitory Concentration (IC50) | >5 uM | N.A. | N.A. | P388 cell | CVCL_7222 | [333] |
| Half Maximal Inhibitory Concentration (IC50) | >5 uM | N.A. | N.A. | L1210 cell | CVCL_0382 | [333] |
| Half Maximal Inhibitory Concentration (IC50) | 5.3 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [412] |
| Half Maximal Inhibitory Concentration (IC50) | 5.63 uM | N.A. | N.A. | K562 cell | CVCL_0004 | [384] |
| Half Maximal Inhibitory Concentration (IC50) | 5.8 uM | N.A. | N.A. | K562 cell | CVCL_0004 | [385] |
| Half Maximal Inhibitory Concentration (IC50) | 5.82 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [413] |
| Half Maximal Inhibitory Concentration (IC50) | 5.898 uM | N.A. | N.A. | A2780 cell | CVCL_0134 | [264] |
| Half Maximal Inhibitory Concentration (IC50) | 5.9 uM | N.A. | N.A. | KB cell | CVCL_0372 | [414] |
| Half Maximal Inhibitory Concentration (IC50) | >6 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [237] |
| Half Maximal Inhibitory Concentration (IC50) | 6.18 uM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [374] |
| Half Maximal Inhibitory Concentration (IC50) | 6.2 uM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [98] |
| Half Maximal Inhibitory Concentration (IC50) | 6.29 uM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [385] |
| Half Maximal Inhibitory Concentration (IC50) | 7.1 uM | N.A. | N.A. | 769-P cell | CVCL_1050 | [347] |
| Half Maximal Inhibitory Concentration (IC50) | 8.1 uM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [234] |
| Half Maximal Inhibitory Concentration (IC50) | 8.2 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [167] |
| Half Maximal Inhibitory Concentration (IC50) | 8.56 uM | N.A. | N.A. | SH-SY5Y cell | CVCL_0019 | [415] |
| Half Maximal Inhibitory Concentration (IC50) | 8.7 uM | N.A. | N.A. | HCT-8 cell | CVCL_2478 | [405] |
| Half Maximal Inhibitory Concentration (IC50) | 9.265 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [416] |
| Half Maximal Inhibitory Concentration (IC50) | 9.4 uM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [6] |
| Half Maximal Inhibitory Concentration (IC50) | 9.49 uM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [241] |
| Half Maximal Inhibitory Concentration (IC50) | 9.8 uM | N.A. | N.A. | KB-V1 cell | CVCL_2089 | [147] |
| Half Maximal Inhibitory Concentration (IC50) | >10 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [417] |
| Half Maximal Inhibitory Concentration (IC50) | <10 uM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [418] |
| Half Maximal Inhibitory Concentration (IC50) | >10 uM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [419] |
| Half Maximal Inhibitory Concentration (IC50) | >10 uM | N.A. | N.A. | HCC1937 cell | CVCL_0290 | [420] |
| Half Maximal Inhibitory Concentration (IC50) | 10.3 uM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [421] |
| Half Maximal Inhibitory Concentration (IC50) | 10.32 uM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [422] |
| Half Maximal Inhibitory Concentration (IC50) | 10.9 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [423] |
| Half Maximal Inhibitory Concentration (IC50) | 11.2 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [424] |
| Half Maximal Inhibitory Concentration (IC50) | 13.05 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [374] |
| Half Maximal Inhibitory Concentration (IC50) | 13.23 uM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [425] |
| Half Maximal Inhibitory Concentration (IC50) | 13.3 uM | N.A. | N.A. | HCT-8 cell | CVCL_2478 | [357] |
| Half Maximal Inhibitory Concentration (IC50) | 17.68 uM | N.A. | N.A. | NCI-ADR-RES cell | CVCL_1452 | [426] |
| Half Maximal Inhibitory Concentration (IC50) | 19.14 uM | N.A. | N.A. | B16-F10 cell | CVCL_0159 | [427] |
| Half Maximal Inhibitory Concentration (IC50) | 19.31 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [428] |
| Half Maximal Inhibitory Concentration (IC50) | 25.12 uM | N.A. | N.A. | A2780/Taxol cell | CVCL_IJ13 | [298] |
| Half Maximal Inhibitory Concentration (IC50) | >30 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [429] |
| Half Maximal Inhibitory Concentration (IC50) | 30.7 uM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [430] |
| Half Maximal Inhibitory Concentration (IC50) | 30.74 uM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [418] |
| Half Maximal Inhibitory Concentration (IC50) | 34.3 uM | N.A. | N.A. | A549/TR cell | CVCL_C5S0 | [431] |
| Half Maximal Inhibitory Concentration (IC50) | 39 uM | N.A. | N.A. | DLD-1 cell | CVCL_0248 | [6] |
| Half Maximal Inhibitory Concentration (IC50) | >50 uM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [311] |
| Half Maximal Inhibitory Concentration (IC50) | 53 uM | N.A. | N.A. | AGS cell | CVCL_0139 | [432] |
| Half Maximal Inhibitory Concentration (IC50) | 54.8 uM | N.A. | N.A. | MCF7/PTX cell | CVCL_C5RS | [385] |
| Half Maximal Inhibitory Concentration (IC50) | 56 uM | N.A. | N.A. | B16 cell | CVCL_F936 | [433] |
| Half Maximal Inhibitory Concentration (IC50) | 56.6 uM | N.A. | N.A. | A2780/Taxol cell | CVCL_IJ13 | [434] |
| Half Maximal Inhibitory Concentration (IC50) | 57.2 uM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [430] |
| Half Maximal Inhibitory Concentration (IC50) | 89.06 uM | N.A. | N.A. | MKN45 cell | CVCL_0434 | [323] |
| Half Maximal Inhibitory Concentration (IC50) | >100 uM | N.A. | N.A. | NCI-ADR-RES cell | CVCL_1452 | [435] |
| Half Maximal Inhibitory Concentration (IC50) | >100 uM | N.A. | N.A. | K562 cell | CVCL_0004 | [436] |
| Half Maximal Inhibitory Concentration (IC50) | 190 uM | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [437] |
| Half Maximal Inhibitory Concentration (IC50) | 400 uM | N.A. | N.A. | KB 3-1 cell | CVCL_2088 | [406] |
| Half Maximal Inhibitory Concentration (IC50) | 854 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [438] |
| Half Maximal Lethal Concentration (IC50) | 3 nM | N.A. | N.A. | CCRF-CEM cell | CVCL_0207 | [439] |
| Half Maximal Lethal Concentration (IC50) | 145 nM | N.A. | N.A. | COLO205 cell | CVCL_F402 | [52] |
| Half Maximal Lethal Concentration (IC50) | >1000 nM | N.A. | N.A. | DMS 114 cell | CVCL_1174 | [52] |
| Half Maximal Lethal Concentration (IC50) | >1000 nM | N.A. | N.A. | SNB-75 cell | CVCL_1706 | [52] |
| Half Maximal Lethal Concentration (IC50) | 79.43 uM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [440] |
| Half Maximal Lethal Concentration (IC50) | >100 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [440] |
| Half Maximal Lethal Concentration (IC50) | >100 uM | N.A. | N.A. | K562 cell | CVCL_0004 | [440] |
| Half Maximal Lethal dose (LD50) | 14 ng/mL | N.A. | N.A. | L1210 cell | CVCL_0382 | [441] |
| Half Maximal Lethal dose (LD50) | 35 uM | N.A. | N.A. | WI-38 cell | CVCL_0579 | [442] |
| Minimum Inhibitory Concentration (MIC) | 5.32 ug/mL | N.A. | N.A. | HeLa cell | CVCL_0030 | [38] |
| Minimum Inhibitory Concentration (MIC) | 39 ng/mL | N.A. | N.A. | A-549 cell | CVCL_0023 | [38] |
| Tumor Growth Inhibition value (TGI) | <30 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [59] |
| Tumor Growth Inhibition value (TGI) | <30 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [59] |
| Tumor Growth Inhibition value (TGI) | 48.7 nM | N.A. | N.A. | OVCAR-3 cell | CVCL_0465 | [52] |
| Tumor Growth Inhibition value (TGI) | 61.2 nM | N.A. | N.A. | DMS 114 cell | CVCL_1174 | [52] |
| Tumor Growth Inhibition value (TGI) | 77.3 nM | N.A. | N.A. | SNB-75 cell | CVCL_1706 | [52] |
| Tumor Growth Inhibition value (TGI) | 80 nM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [59] |
| Tumor Growth Inhibition value (TGI) | 89.9 nM | N.A. | N.A. | SF539 cell | CVCL_1691 | [52] |
| Tumor Growth Inhibition value (TGI) | 126 nM | N.A. | N.A. | SNB-75 cell | CVCL_1706 | [58] |
| Tumor Growth Inhibition value (TGI) | 126 nM | N.A. | N.A. | HCC 2998 cell | CVCL_1266 | [58] |
| Tumor Growth Inhibition value (TGI) | 251 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [58] |
| Tumor Growth Inhibition value (TGI) | 794 nM | N.A. | N.A. | DU145 cell | CVCL_0105 | [53] |
| Tumor Growth Inhibition value (TGI) | >1000 nM | N.A. | N.A. | HOP-62 cell | CVCL_1285 | [52] |
| Tumor Growth Inhibition value (TGI) | >1000 nM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [52] |
| Tumor Growth Inhibition value (TGI) | 5.012 uM | N.A. | N.A. | RPMI-8226 cell | CVCL_7353 | [58] |
| Tumor Growth Inhibition value (TGI) | 10 uM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [58] |
| Tumor Growth Inhibition value (TGI) | 12.59 uM | N.A. | N.A. | ACHN cell | CVCL_1067 | [53] |
| Tumor Growth Inhibition value (TGI) | 12.59 uM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [440] |
| Tumor Growth Inhibition value (TGI) | 15.85 uM | N.A. | N.A. | KM12 cell | CVCL_1331 | [53] |
| Tumor Growth Inhibition value (TGI) | 15.85 uM | N.A. | N.A. | K562 cell | CVCL_0004 | [440] |
| Tumor Growth Inhibition value (TGI) | 25.11 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [53] |
| Tumor Growth Inhibition value (TGI) | 25.12 uM | N.A. | N.A. | UACC-257 cell | CVCL_1779 | [58] |
| Tumor Growth Inhibition value (TGI) | 79.43 uM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [440] |
Each Peptide-drug Conjugate Related to This Drug
Full Information of The Activity Data of The PDC(s) Related to This Drug
ANG1005 [Phase 3]
Identified from the Human Clinical Data
| Experiment 1 Reporting the Activity Data of This PDC | [443] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Objective response rate (ORR) | 15% | |||
| Patients Enrolled |
Adult patients with measurable recurrent brain metastases from breast cancer.
|
||||
| Administration Time | Every 3 weeks | ||||
| Administration Dosage | 600 mg/m2 | ||||
| MOA of PDC |
Because LRP1 is also expressed on tumor cells in both CNS and systemic metastases, ANG1005 gains entry via LRP1 mediated endocytosis, where paclitaxel is cleaved from the peptide backbone by lysosomal esterases.
|
||||
| Description |
On the basis of the CNS tumor response assessment, performed by local investigators, there were nine (15%) evaluable patients with PR including five (8%) confirmed PR (to confirm PR, it was required that the response was sustained for ≥4 weeks), and 32 (53%) evaluable patients with SD, resulting in an overall iORR of 15% and iCBR of 68%.
|
||||
| Experiment 2 Reporting the Activity Data of This PDC | [443] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Objective response rate (ORR) | 29% | |||
| Patients Enrolled |
Patients with leptomeningeal carcinomatosis.
|
||||
| Administration Time | Every 3 weeks | ||||
| Administration Dosage | 600 mg/m2 | ||||
| MOA of PDC |
Because LRP1 is also expressed on tumor cells in both CNS and systemic metastases, ANG1005 gains entry via LRP1 mediated endocytosis, where paclitaxel is cleaved from the peptide backbone by lysosomal esterases.
|
||||
| Description |
Investigator determined ORR was 29% and the iCBR was 67%.
|
||||
| Experiment 3 Reporting the Activity Data of This PDC | [443] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Median progression-free survival (mPFS) | 2.8 months | |||
| Patients Enrolled |
Patients with leptomeningeal carcinomatosis.
|
||||
| Administration Time | Every 3 weeks | ||||
| Administration Dosage | 600 mg/m2 | ||||
| MOA of PDC |
Because LRP1 is also expressed on tumor cells in both CNS and systemic metastases, ANG1005 gains entry via LRP1 mediated endocytosis, where paclitaxel is cleaved from the peptide backbone by lysosomal esterases.
|
||||
| Description |
The investigator determined intracranial median PFS was 2.8 months and the 3-month PFS rate was 54% (Table 3). Median duration of response was 18 weeks (7.3-26.3).
|
||||
| Experiment 4 Reporting the Activity Data of This PDC | [443] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Median progression-free survival (mPFS) | 12.1 weeks | |||
| Patients Enrolled |
Adult patients with measurable recurrent brain metastases from breast cancer.
|
||||
| Administration Time | Every 3 weeks | ||||
| Administration Dosage | 600 mg/m2 | ||||
| MOA of PDC |
Because LRP1 is also expressed on tumor cells in both CNS and systemic metastases, ANG1005 gains entry via LRP1 mediated endocytosis, where paclitaxel is cleaved from the peptide backbone by lysosomal esterases.
|
||||
| Description |
Investigator assessments resulted in median intracranial PFS of 2.8 months and the 3-month intracranial PFS rate was 52%.
|
||||
| Experiment 5 Reporting the Activity Data of This PDC | [443] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Median duration of response | 18 weeks | |||
| Patients Enrolled |
Patients with leptomeningeal carcinomatosis.
|
||||
| Administration Time | Every 3 weeks | ||||
| Administration Dosage | 600 mg/m2 | ||||
| MOA of PDC |
Because LRP1 is also expressed on tumor cells in both CNS and systemic metastases, ANG1005 gains entry via LRP1 mediated endocytosis, where paclitaxel is cleaved from the peptide backbone by lysosomal esterases.
|
||||
| Description |
The investigator determined intracranial median PFS was 2.8 months and the 3-month PFS rate was 54% (Table 3). Median duration of response was 18 weeks (7.3-26.3).
|
||||
| Experiment 6 Reporting the Activity Data of This PDC | [443] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Clinical benefit rate (CBR) | 67% | |||
| Patients Enrolled |
Patients with leptomeningeal carcinomatosis.
|
||||
| Administration Time | Every 3 weeks | ||||
| Administration Dosage | 600 mg/m2 | ||||
| MOA of PDC |
Because LRP1 is also expressed on tumor cells in both CNS and systemic metastases, ANG1005 gains entry via LRP1 mediated endocytosis, where paclitaxel is cleaved from the peptide backbone by lysosomal esterases.
|
||||
| Description |
Investigator determined ORR was 29% and the iCBR was 67%.
|
||||
| Experiment 7 Reporting the Activity Data of This PDC | [443] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Clinical benefit rate (CBR) | 68% | |||
| Patients Enrolled |
Adult patients with measurable recurrent brain metastases from breast cancer.
|
||||
| Administration Time | Every 3 weeks | ||||
| Administration Dosage | 600 mg/m2 | ||||
| MOA of PDC |
Because LRP1 is also expressed on tumor cells in both CNS and systemic metastases, ANG1005 gains entry via LRP1 mediated endocytosis, where paclitaxel is cleaved from the peptide backbone by lysosomal esterases.
|
||||
| Description |
On the basis of the CNS tumor response assessment, performed by local investigators, there were nine (15%) evaluable patients with PR including five (8%) confirmed PR (to confirm PR, it was required that the response was sustained for ≥4 weeks), and 32 (53%) evaluable patients with SD, resulting in an overall iORR of 15% and iCBR of 68%.
|
||||
| Experiment 8 Reporting the Activity Data of This PDC | [443] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | 6-month progression-free survival rate | 18.70% | |||
| Patients Enrolled |
Adult patients with measurable recurrent brain metastases from breast cancer.
|
||||
| Administration Time | Every 3 weeks | ||||
| Administration Dosage | 600 mg/m2 | ||||
| MOA of PDC |
Because LRP1 is also expressed on tumor cells in both CNS and systemic metastases, ANG1005 gains entry via LRP1 mediated endocytosis, where paclitaxel is cleaved from the peptide backbone by lysosomal esterases.
|
||||
| Description |
Investigator assessments resulted in median intracranial PFS of 2.8 months and the 3-month intracranial PFS rate was 52%.
|
||||
| Experiment 9 Reporting the Activity Data of This PDC | [443] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | 3-month progression-free survival rate | 52.00% | |||
| Patients Enrolled |
Adult patients with measurable recurrent brain metastases from breast cancer.
|
||||
| Administration Time | Every 3 weeks | ||||
| Administration Dosage | 600 mg/m2 | ||||
| MOA of PDC |
Because LRP1 is also expressed on tumor cells in both CNS and systemic metastases, ANG1005 gains entry via LRP1 mediated endocytosis, where paclitaxel is cleaved from the peptide backbone by lysosomal esterases.
|
||||
| Description |
Investigator assessments resulted in median intracranial PFS of 2.8 months and the 3-month intracranial PFS rate was 52%.
|
||||
| Experiment 10 Reporting the Activity Data of This PDC | [443] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | 3-month progression-free survival rate | 54.00% | |||
| Patients Enrolled |
Patients with leptomeningeal carcinomatosis.
|
||||
| Administration Time | Every 3 weeks | ||||
| Administration Dosage | 600 mg/m2 | ||||
| MOA of PDC |
Because LRP1 is also expressed on tumor cells in both CNS and systemic metastases, ANG1005 gains entry via LRP1 mediated endocytosis, where paclitaxel is cleaved from the peptide backbone by lysosomal esterases.
|
||||
| Description |
The investigator determined intracranial median PFS was 2.8 months and the 3-month PFS rate was 54% (Table 3). Median duration of response was 18 weeks (7.3-26.3).
|
||||
SBI1301 [Preclinical]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [444] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) | 10% | |||
| Evaluation Method | Tumor volume detection | ||||
| Administration Time | 54 days | ||||
| Administration Dosage | 0.3 mg/kg | ||||
| Description |
In xenografted prostate cancer in mice, only 3 treatments over 12 days showed complete tumor regression. At the highest dose tested, there were no obvious symptoms of toxicity. After 60 days of observation, the tumor did not regrow.
|
||||
| In Vivo Model | PC-3 mouse xenograft with prostate cancer | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [444] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) | 75% | |||
| Evaluation Method | Tumor volume detection | ||||
| Administration Time | 54 days | ||||
| Administration Dosage | 0.6 mg/kg | ||||
| Description |
In xenografted prostate cancer in mice, only 3 treatments over 12 days showed complete tumor regression. At the highest dose tested, there were no obvious symptoms of toxicity. After 60 days of observation, the tumor did not regrow.
|
||||
| In Vivo Model | PC-3 mouse xenograft with prostate cancer | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [444] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) | 100% | |||
| Evaluation Method | Tumor volume detection | ||||
| Administration Time | 54 days | ||||
| Administration Dosage | 1.2 mg/kg | ||||
| Description |
In xenografted prostate cancer in mice, only 3 treatments over 12 days showed complete tumor regression. At the highest dose tested, there were no obvious symptoms of toxicity. After 60 days of observation, the tumor did not regrow.
|
||||
| In Vivo Model | PC-3 mouse xenograft with prostate cancer | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [444] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) | 100% | |||
| Evaluation Method | Tumor volume detection | ||||
| Administration Time | 54 days | ||||
| Administration Dosage | 2.4 mg/kg | ||||
| Description |
In xenografted prostate cancer in mice, only 3 treatments over 12 days showed complete tumor regression. At the highest dose tested, there were no obvious symptoms of toxicity. After 60 days of observation, the tumor did not regrow.
|
||||
| In Vivo Model | PC-3 mouse xenograft with prostate cancer | ||||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [444] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 2.3 nM | |||
| Description |
In xenografted prostate cancer in mice, only 3 treatments over 12 days showed complete tumor regression. At the highest dose tested, there were no obvious symptoms of toxicity. After 60 days of observation, the tumor did not regrow.
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | BxPC-3 cell | CVCL_0186 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [444] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 2.4 nM | |||
| Description |
In xenografted prostate cancer in mice, only 3 treatments over 12 days showed complete tumor regression. At the highest dose tested, there were no obvious symptoms of toxicity. After 60 days of observation, the tumor did not regrow.
|
||||
| In Vitro Model | Prostate carcinoma | PC-3 cell | CVCL_0035 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [444] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 2.6 nM | |||
| Description |
In xenografted prostate cancer in mice, only 3 treatments over 12 days showed complete tumor regression. At the highest dose tested, there were no obvious symptoms of toxicity. After 60 days of observation, the tumor did not regrow.
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | MIA PaCa-2 cell | CVCL_0428 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [444] | ||||
| Indication | Uterine corpus sarcoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 5.7 nM | |||
| Description |
In xenografted prostate cancer in mice, only 3 treatments over 12 days showed complete tumor regression. At the highest dose tested, there were no obvious symptoms of toxicity. After 60 days of observation, the tumor did not regrow.
|
||||
| In Vitro Model | Uterine corpus sarcoma | MES-SA cell | CVCL_1404 | ||
P-(A5G27scrm)-PTX [Investigative]
Identified from the Human Clinical Data
| Experiment 1 Reporting the Activity Data of This PDC | [445] | ||||
| Indication | Metastatic Tumor | ||||
| Efficacy Data | Median overall survival (mOS) | 40.5 days | |||
| Administration Dosage | 15 mg/kg PTX equivalent dose | ||||
| Description |
The median survival of mice in the P-(A5G27)-PTX treatment group was longer than in P-(A5G27scrm)-PTX, P-(A5G27), and free PTX-treated groups (48.50 vs 40.5, 43, and 45.5, respectively); however, differences were nonsignificant (Figure 6B).
|
||||
| In Vivo Model | 4T1 tumor-bearing mice. | ||||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [445] | ||||
| Indication | Metastatic Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.221 ± 0.055 µM | |||
| Administration Time | 72 h | ||||
| Description |
P-(A5G27)-PTX was ˜4-fold more toxic than the nontargeted P-(A5G27scrm)-PTX copolymer, suggesting a faster (receptor-mediated) internalization (Figure 4).
|
||||
| In Vitro Model | Mammary carcinoma | 4T1 cell | CVCL_0125 | ||
P-(A5G27)-PTX [Investigative]
Identified from the Human Clinical Data
| Experiment 1 Reporting the Activity Data of This PDC | [445] | ||||
| Indication | Metastatic Tumor | ||||
| Efficacy Data | Median overall survival (mOS) | 48.50 days | |||
| Administration Dosage | 15 mg/kg PTX equivalent dose | ||||
| Description |
The median survival of mice in the P-(A5G27)-PTX treatment group was longer than in P-(A5G27scrm)-PTX, P-(A5G27), and free PTX-treated groups (48.50 vs 40.5, 43, and 45.5, respectively); however, differences were nonsignificant (Figure 6B).
|
||||
| In Vivo Model | 4T1 tumor-bearing mice. | ||||
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [445] | ||||
| Indication | Metastatic Tumor | ||||
| Efficacy Data | Survival rate | 40% | |||
| Administration Time | 7 days | ||||
| Administration Dosage | 15 mg/kg PTX equivalent dose | ||||
| Description |
P-(A5G27)-PTX prolonged mice survival in 7 and 20% relative to the survival rates of free PTX and nontreated mice, respectively (Figure 5).
|
||||
| In Vivo Model | B16-F10 melanoma tumor-bearing mice model. | ||||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [445] | ||||
| Indication | Metastatic Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.058 ± 0.015 µM | |||
| Administration Time | 72 h | ||||
| Description |
P-(A5G27)-PTX was ˜4-fold more toxic than the nontargeted P-(A5G27scrm)-PTX copolymer, suggesting a faster (receptor-mediated) internalization (Figure 4).
|
||||
| In Vitro Model | Mammary carcinoma | 4T1 cell | CVCL_0125 | ||
FA-P7-PTX [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [446] | ||||
| Indication | Hepatoma | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 0.00% | |||
| Administration Time | 4 days | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
To study the anticancer activity of FA-P7-PTX in vivo, we performed tumor-bearing mice model with H22cells by administering once every two days peritumoral injection of FA-P7-PTX (12 μmol/kg), PTX (12 μmol/kg, as the positive control), or 0.9% saline as the negative control for 2 weeks. Compared with control group, the tumor volumes of the FA-P7-PTX group were dramatically reduced by 69% with no significant variation in mouse body weight. Meanwhile FA-P7-PTX exhibited stronger inhibitory effects on tumor volume compared with PTX (69% versus 49%). The result confirmed that FA-P7-PTX possessed higher potency in slowing the growth of solid tumors.
Click to Show/Hide
|
||||
| In Vivo Model | Tumor-bearing mice model with H22 cells. | ||||
| In Vitro Model | Hepatoma | H22 cell | CVCL_H613 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [446] | ||||
| Indication | Hepatoma | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 16.70% | |||
| Administration Time | 6 days | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
To study the anticancer activity of FA-P7-PTX in vivo, we performed tumor-bearing mice model with H22cells by administering once every two days peritumoral injection of FA-P7-PTX (12 μmol/kg), PTX (12 μmol/kg, as the positive control), or 0.9% saline as the negative control for 2 weeks. Compared with control group, the tumor volumes of the FA-P7-PTX group were dramatically reduced by 69% with no significant variation in mouse body weight. Meanwhile FA-P7-PTX exhibited stronger inhibitory effects on tumor volume compared with PTX (69% versus 49%). The result confirmed that FA-P7-PTX possessed higher potency in slowing the growth of solid tumors.
Click to Show/Hide
|
||||
| In Vivo Model | Tumor-bearing mice model with H22 cells. | ||||
| In Vitro Model | Hepatoma | H22 cell | CVCL_H613 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [446] | ||||
| Indication | Hepatoma | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 38.90% | |||
| Administration Time | 8 days | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
To study the anticancer activity of FA-P7-PTX in vivo, we performed tumor-bearing mice model with H22cells by administering once every two days peritumoral injection of FA-P7-PTX (12 μmol/kg), PTX (12 μmol/kg, as the positive control), or 0.9% saline as the negative control for 2 weeks. Compared with control group, the tumor volumes of the FA-P7-PTX group were dramatically reduced by 69% with no significant variation in mouse body weight. Meanwhile FA-P7-PTX exhibited stronger inhibitory effects on tumor volume compared with PTX (69% versus 49%). The result confirmed that FA-P7-PTX possessed higher potency in slowing the growth of solid tumors.
Click to Show/Hide
|
||||
| In Vivo Model | Tumor-bearing mice model with H22 cells. | ||||
| In Vitro Model | Hepatoma | H22 cell | CVCL_H613 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [446] | ||||
| Indication | Hepatoma | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 50.00% | |||
| Administration Time | 10 days | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
To study the anticancer activity of FA-P7-PTX in vivo, we performed tumor-bearing mice model with H22cells by administering once every two days peritumoral injection of FA-P7-PTX (12 μmol/kg), PTX (12 μmol/kg, as the positive control), or 0.9% saline as the negative control for 2 weeks. Compared with control group, the tumor volumes of the FA-P7-PTX group were dramatically reduced by 69% with no significant variation in mouse body weight. Meanwhile FA-P7-PTX exhibited stronger inhibitory effects on tumor volume compared with PTX (69% versus 49%). The result confirmed that FA-P7-PTX possessed higher potency in slowing the growth of solid tumors.
Click to Show/Hide
|
||||
| In Vivo Model | Tumor-bearing mice model with H22 cells. | ||||
| In Vitro Model | Hepatoma | H22 cell | CVCL_H613 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [446] | ||||
| Indication | Hepatoma | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 63.00% | |||
| Administration Time | 12 days | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
To study the anticancer activity of FA-P7-PTX in vivo, we performed tumor-bearing mice model with H22cells by administering once every two days peritumoral injection of FA-P7-PTX (12 μmol/kg), PTX (12 μmol/kg, as the positive control), or 0.9% saline as the negative control for 2 weeks. Compared with control group, the tumor volumes of the FA-P7-PTX group were dramatically reduced by 69% with no significant variation in mouse body weight. Meanwhile FA-P7-PTX exhibited stronger inhibitory effects on tumor volume compared with PTX (69% versus 49%). The result confirmed that FA-P7-PTX possessed higher potency in slowing the growth of solid tumors.
Click to Show/Hide
|
||||
| In Vivo Model | Tumor-bearing mice model with H22 cells. | ||||
| In Vitro Model | Hepatoma | H22 cell | CVCL_H613 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [446] | ||||
| Indication | Hepatoma | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 65.20% | |||
| Administration Time | 16 days | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
To study the anticancer activity of FA-P7-PTX in vivo, we performed tumor-bearing mice model with H22cells by administering once every two days peritumoral injection of FA-P7-PTX (12 μmol/kg), PTX (12 μmol/kg, as the positive control), or 0.9% saline as the negative control for 2 weeks. Compared with control group, the tumor volumes of the FA-P7-PTX group were dramatically reduced by 69% with no significant variation in mouse body weight. Meanwhile FA-P7-PTX exhibited stronger inhibitory effects on tumor volume compared with PTX (69% versus 49%). The result confirmed that FA-P7-PTX possessed higher potency in slowing the growth of solid tumors.
Click to Show/Hide
|
||||
| In Vivo Model | Tumor-bearing mice model with H22 cells. | ||||
| In Vitro Model | Hepatoma | H22 cell | CVCL_H613 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [446] | ||||
| Indication | Hepatoma | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 66.90% | |||
| Administration Time | 14 days | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
To study the anticancer activity of FA-P7-PTX in vivo, we performed tumor-bearing mice model with H22cells by administering once every two days peritumoral injection of FA-P7-PTX (12 μmol/kg), PTX (12 μmol/kg, as the positive control), or 0.9% saline as the negative control for 2 weeks. Compared with control group, the tumor volumes of the FA-P7-PTX group were dramatically reduced by 69% with no significant variation in mouse body weight. Meanwhile FA-P7-PTX exhibited stronger inhibitory effects on tumor volume compared with PTX (69% versus 49%). The result confirmed that FA-P7-PTX possessed higher potency in slowing the growth of solid tumors.
Click to Show/Hide
|
||||
| In Vivo Model | Tumor-bearing mice model with H22 cells. | ||||
| In Vitro Model | Hepatoma | H22 cell | CVCL_H613 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [446] | ||||
| Indication | Hepatoma | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 67.30% | |||
| Administration Time | 18 days | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
To study the anticancer activity of FA-P7-PTX in vivo, we performed tumor-bearing mice model with H22cells by administering once every two days peritumoral injection of FA-P7-PTX (12 μmol/kg), PTX (12 μmol/kg, as the positive control), or 0.9% saline as the negative control for 2 weeks. Compared with control group, the tumor volumes of the FA-P7-PTX group were dramatically reduced by 69% with no significant variation in mouse body weight. Meanwhile FA-P7-PTX exhibited stronger inhibitory effects on tumor volume compared with PTX (69% versus 49%). The result confirmed that FA-P7-PTX possessed higher potency in slowing the growth of solid tumors.
Click to Show/Hide
|
||||
| In Vivo Model | Tumor-bearing mice model with H22 cells. | ||||
| In Vitro Model | Hepatoma | H22 cell | CVCL_H613 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [446] | ||||
| Indication | Hepatoma | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 69.10% | |||
| Administration Time | 20 days | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
To study the anticancer activity of FA-P7-PTX in vivo, we performed tumor-bearing mice model with H22cells by administering once every two days peritumoral injection of FA-P7-PTX (12 μmol/kg), PTX (12 μmol/kg, as the positive control), or 0.9% saline as the negative control for 2 weeks. Compared with control group, the tumor volumes of the FA-P7-PTX group were dramatically reduced by 69% with no significant variation in mouse body weight. Meanwhile FA-P7-PTX exhibited stronger inhibitory effects on tumor volume compared with PTX (69% versus 49%). The result confirmed that FA-P7-PTX possessed higher potency in slowing the growth of solid tumors.
Click to Show/Hide
|
||||
| In Vivo Model | Tumor-bearing mice model with H22 cells. | ||||
| In Vitro Model | Hepatoma | H22 cell | CVCL_H613 | ||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [446] | ||||
| Indication | Invasive breast carcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 1.39 ± 0.12 μM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [446] | ||||
| Indication | Ovarian endometrioid adenocarcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 1.42 ± 0.08 μM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [446] | ||||
| Indication | Invasive ductal carcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 2.92 ± 0.2 μM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive ductal carcinoma | MCF7/PTX cell | CVCL_C5RS | ||
| Experiment 4 Reporting the Activity Data of This PDC | [446] | ||||
| Indication | Chronic myeloid leukemia | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 3.85 ± 0.9 μM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Chronic myeloid leukemia | K562 cell | CVCL_0004 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [446] | ||||
| Indication | Ovarian serous cystadenocarcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 5.49 ± 0.36 μM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [446] | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 45.21 ± 1.79 μM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Human umbilical vein endothelial cells | Homo sapiens | ||
PTX-SM-TAR [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) | 43.24% | |||
| Administration Time | 16 d | ||||
| Administration Dosage | 10.6 mg/kg | ||||
| MOA of PDC |
Triple-negative breast cancer (TNBC) is an extremely aggressive subtype associated with a poor prognosis. At present, the treatment for TNBC mainly relies on surgery and traditional chemotherapy. As a key component in the standard treatment of TNBC, paclitaxel (PTX) effectively inhibits the growth and proliferation of tumor cells. However, the application of PTX in clinical treatment is limited due to its inherent hydrophobicity, weak penetrability, nonspecific accumulation, and side effects. To counter these problems, we constructed a novel PTX conjugate based on the peptide-drug conjugates (PDCs) strategy. In this PTX conjugate, a novel fused peptide TAR consisting of a tumor-targeting peptide, A7R, and a cell-penetrating peptide, TAT, is used to modify PTX. After modification, this conjugate is named PTX-SM-TAR, which is expected to improve the specificity and penetrability of PTX at the tumor site. Depending on hydrophilic TAR peptide and hydrophobic PTX, PTX-SM-TAR can self-assemble into nanoparticles and improve the water solubility of PTX. In terms of linkage, the acid- and esterase-sensitive ester bond was used as the linking bond, with which PTX-SM-TAR NPs could remain stable in the physiological environment, whereas PTX-SM-TAR NPs could be broken and PTX be released at the tumor site. A cell uptake assay showed that PTX-SM-TAR NPs were receptor-targeting and could mediate endocytosis by binding to NRP-1. The vascular barrier, transcellular migration, and tumor spheroids experiments showed that PTX-SM-TAR NPs exhibit great transvascular transport and tumor penetration ability. In vivo experiments, PTX-SM-TAR NPs showed higher antitumor effects than PTX. As a result, PTX-SM-TAR NPs may overcome the shortcomings of PTX and present a new transcytosable and targeted delivery system for PTX in TNBC treatment.
Click to Show/Hide
|
||||
| Description |
The mice TNBC 4T1-mCherry-Luc model was constructed by subcutaneous inoculation to study the anti-tumor effect of PTX-SM-TAR NPs in vivo. 4T1-mCherry-Luc cells reacted with D-luciferin potassium salt to produce bioluminescence, which was determined by an IVIS spectrum imaging system. The intensity of the fluorescence signal is related to tumor size, hence tumor growth in mice can be monitored in real-time. As shown in Figure 9a, the tumor growth rate of the PTX-SM-TAR NPs group was lower than that of the NS, TAR, and PTX groups. At the end of the experiment, the tumor tissue was weighed, and the results were consistent with the trend of fluorescence intensity. The tumor inhibition rate was 43.24% in the PTX-SM-TAR NPs, 28.47% in the PTX, and 7.81% in the TAR. The tumor inhibitory effect of the PTX-SM-TAR NPs group was stronger than that of the PTX group, and the difference was significant.
Click to Show/Hide
|
||||
| In Vivo Model | Female BALB/c mice 4T1-mCherry-Luc cells xenograft model. | ||||
| In Vitro Model | Malignant neoplasms of the mouse mammary gland | 4T1-mCherry-Luc cell | CVCL_C8UZ | ||
BPP-PTX [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [447] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 64.00% | |||
| Administration Time | 30 days | ||||
| Administration Dosage | 2.4 μmol/kg | ||||
| MOA of PDC |
Here, we report the design, synthesis, and evaluation-in vitro and in vivo-of a novel PDC, namely BPP-PTX, whereby PTX is conjugated to one member of BPPs, Bj-BPP-9a (teprotide), via a succinyl linker. The targeting moiety was carefully selected based on previous studies. It was similarly employed with a nanoparticle carrier in vivo and was shown to modulate improved drug accumulation at the tumor site, thereby curbing tumor growth and extending the lives of tumor-bearing mice. In this study, we demonstrate for the first time that BPP-PTX functions through BPPs cognate receptor, ACE. ACE was overexpressed in TNBC cell lines but not in the receptor-positive cell line. BPP, as part of BPP-PTX, bound ACE and mediated its selective cytotoxic action through this receptor in ACE-positive TNBC cells. Furthermore, BPP-PTX demonstrated improved biodistribution, therapeutic activity, and better safety profile in vivo. These results advocate the significance of BPP-PTX as a suitable tumor-targeting PDC, strongly warranting further refinement and investigations for TNBC.
Click to Show/Hide
|
||||
| Description |
To test the in vivo efficacy and toxicity of BPP-PTX, an orthotopic mouse model was established. ACE-positive MDA-MB-468 cells were injected orthotopically into the mammary fat pad of female nude mice. After the inoculated tumors reached a mean volume of 100 mm3, they were randomly divided into four groups to ensure that the mean tumor volume was evaluated and treated for 28 days with PBS, plain PTX (2.4 μmol/kg, equivalent to 2.0 mg/kg), low-dose BPP-PTX (2.4 μmol/kg, equivalent to 4.9 mg/kg), and high-dose BPP-PTX (9.6 μmol/kg, equivalent to 19.6 mg/kg) every 4 days by i.p. injection. Tumor growth was assessed by measuring tumor volume every 4 days. On the fourth day of testing after the first injection, there was no significant difference in tumor volume compared with control and drug-treated groups. In the subsequent treatments, the tumors of the control mice grew significantly faster than those of mice treated with plain PTX and BPP-PTX. On day 28, mice treated with low-dose BPP-PTX showed an approximately 15% reduction in mean tumor volume compared with mice treated with plain PTX and a 54% reduction compared to controls (PBS). Meanwhile, the mean tumor volumes of mice treated with high-dose BPP-PTX were reduced by 62% compared with control animals and by approximately 30% compared to animals treated with plain PTX. Consistently, the mean tumor weight of mice with low-dose BPP-PTX treatment was 0.20 g (0.17, 0.24), which is lower than that of mice treated with plain PTX [0.26 g (0.21, 0.31)] and control mice [0.43 g (0.37, 0.49)]. The tumor weight of mice treated with high-dose BPP-PTX was significantly lower than that of the control and plain PTX-treated groups, with a mean tumor weight of only 0.16 g (0.14, 0.19). These results suggested that BPP-PTX has good tumor-suppression efficacy in vivo, even better than that of plain PTX.
Click to Show/Hide
|
||||
| In Vivo Model | TNBC nude mouse orthotopic transplantation tumor model. | ||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-468 cell | CVCL_0419 | ||
| Half life period | 5.82 h | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [447] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 74.00% | |||
| Administration Time | 30 days | ||||
| Administration Dosage | 9.6 μmol/kg | ||||
| MOA of PDC |
Here, we report the design, synthesis, and evaluation-in vitro and in vivo-of a novel PDC, namely BPP-PTX, whereby PTX is conjugated to one member of BPPs, Bj-BPP-9a (teprotide), via a succinyl linker. The targeting moiety was carefully selected based on previous studies. It was similarly employed with a nanoparticle carrier in vivo and was shown to modulate improved drug accumulation at the tumor site, thereby curbing tumor growth and extending the lives of tumor-bearing mice. In this study, we demonstrate for the first time that BPP-PTX functions through BPPs cognate receptor, ACE. ACE was overexpressed in TNBC cell lines but not in the receptor-positive cell line. BPP, as part of BPP-PTX, bound ACE and mediated its selective cytotoxic action through this receptor in ACE-positive TNBC cells. Furthermore, BPP-PTX demonstrated improved biodistribution, therapeutic activity, and better safety profile in vivo. These results advocate the significance of BPP-PTX as a suitable tumor-targeting PDC, strongly warranting further refinement and investigations for TNBC.
Click to Show/Hide
|
||||
| Description |
To test the in vivo efficacy and toxicity of BPP-PTX, an orthotopic mouse model was established. ACE-positive MDA-MB-468 cells were injected orthotopically into the mammary fat pad of female nude mice. After the inoculated tumors reached a mean volume of 100 mm3, they were randomly divided into four groups to ensure that the mean tumor volume was evaluated and treated for 28 days with PBS, plain PTX (2.4 μmol/kg, equivalent to 2.0 mg/kg), low-dose BPP-PTX (2.4 μmol/kg, equivalent to 4.9 mg/kg), and high-dose BPP-PTX (9.6 μmol/kg, equivalent to 19.6 mg/kg) every 4 days by i.p. injection. Tumor growth was assessed by measuring tumor volume every 4 days. On the fourth day of testing after the first injection, there was no significant difference in tumor volume compared with control and drug-treated groups. In the subsequent treatments, the tumors of the control mice grew significantly faster than those of mice treated with plain PTX and BPP-PTX. On day 28, mice treated with low-dose BPP-PTX showed an approximately 15% reduction in mean tumor volume compared with mice treated with plain PTX and a 54% reduction compared to controls (PBS). Meanwhile, the mean tumor volumes of mice treated with high-dose BPP-PTX were reduced by 62% compared with control animals and by approximately 30% compared to animals treated with plain PTX. Consistently, the mean tumor weight of mice with low-dose BPP-PTX treatment was 0.20 g (0.17, 0.24), which is lower than that of mice treated with plain PTX [0.26 g (0.21, 0.31)] and control mice [0.43 g (0.37, 0.49)]. The tumor weight of mice treated with high-dose BPP-PTX was significantly lower than that of the control and plain PTX-treated groups, with a mean tumor weight of only 0.16 g (0.14, 0.19). These results suggested that BPP-PTX has good tumor-suppression efficacy in vivo, even better than that of plain PTX.
Click to Show/Hide
|
||||
| In Vivo Model | TNBC nude mouse orthotopic transplantation tumor model. | ||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-468 cell | CVCL_0419 | ||
| Half life period | 5.82 h | ||||
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [447] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Weight loss rate | 1.90% | |||
| Administration Dosage | 60 mg/kg | ||||
| MOA of PDC |
Here, we report the design, synthesis, and evaluation-in vitro and in vivo-of a novel PDC, namely BPP-PTX, whereby PTX is conjugated to one member of BPPs, Bj-BPP-9a (teprotide), via a succinyl linker. The targeting moiety was carefully selected based on previous studies. It was similarly employed with a nanoparticle carrier in vivo and was shown to modulate improved drug accumulation at the tumor site, thereby curbing tumor growth and extending the lives of tumor-bearing mice. In this study, we demonstrate for the first time that BPP-PTX functions through BPPs cognate receptor, ACE. ACE was overexpressed in TNBC cell lines but not in the receptor-positive cell line. BPP, as part of BPP-PTX, bound ACE and mediated its selective cytotoxic action through this receptor in ACE-positive TNBC cells. Furthermore, BPP-PTX demonstrated improved biodistribution, therapeutic activity, and better safety profile in vivo. These results advocate the significance of BPP-PTX as a suitable tumor-targeting PDC, strongly warranting further refinement and investigations for TNBC.
Click to Show/Hide
|
||||
| Description |
After obtaining positive in vitro results, BPP-PTX was then tested in vivo. To test the in vivo toxicity of the drug, the maximum tolerated dose (MTD) of BPP-PTX in Balb/c mice was determined. The single-dose MTD (acute toxicity) of plain PTX was 20 mg/kg (equivalent to 23.4 μmol/kg), similar to previous findings in the literature, while the single-dose MTD of BPP-PTX was 100 mg/kg (equivalent to 48.7 μmol/kg). The weight loss was more severe as the drug dose increased. However, the acute weight loss caused by high dose was temporary, with the body weight recovering to baseline levels within 15 days. Then, we evaluated the plasma pharmacokinetics profiles of plain PTX and BPP-PTX. Mice xenografted with MDA-MB-468 received intraperitoneal (i.p.) injections of 15 mg/kg PTX (equivalent to 17.6 μmol/kg) or 36.1 mg/kg BPP-PTX (equivalent to 17.6 μmol/kg). The plasma PTX concentration in the plain PTX-treated group reached peak levels at 1 h and then decreased rapidly, and it was quickly removed from the circulating system. In contrast, the plasma PTX concentration in the BPP-PTX-treated group remained high for an extended period after 1 h, suggesting that BPP-PTX could have a lower release rate in circulation.
Click to Show/Hide
|
||||
| In Vivo Model | Female BALB/c mice. | ||||
| Half life period | 5.82 h | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [447] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Weight loss rate | 4.50% | |||
| Administration Dosage | 80 mg/kg | ||||
| MOA of PDC |
Here, we report the design, synthesis, and evaluation-in vitro and in vivo-of a novel PDC, namely BPP-PTX, whereby PTX is conjugated to one member of BPPs, Bj-BPP-9a (teprotide), via a succinyl linker. The targeting moiety was carefully selected based on previous studies. It was similarly employed with a nanoparticle carrier in vivo and was shown to modulate improved drug accumulation at the tumor site, thereby curbing tumor growth and extending the lives of tumor-bearing mice. In this study, we demonstrate for the first time that BPP-PTX functions through BPPs cognate receptor, ACE. ACE was overexpressed in TNBC cell lines but not in the receptor-positive cell line. BPP, as part of BPP-PTX, bound ACE and mediated its selective cytotoxic action through this receptor in ACE-positive TNBC cells. Furthermore, BPP-PTX demonstrated improved biodistribution, therapeutic activity, and better safety profile in vivo. These results advocate the significance of BPP-PTX as a suitable tumor-targeting PDC, strongly warranting further refinement and investigations for TNBC.
Click to Show/Hide
|
||||
| Description |
After obtaining positive in vitro results, BPP-PTX was then tested in vivo. To test the in vivo toxicity of the drug, the maximum tolerated dose (MTD) of BPP-PTX in Balb/c mice was determined. The single-dose MTD (acute toxicity) of plain PTX was 20 mg/kg (equivalent to 23.4 μmol/kg), similar to previous findings in the literature, while the single-dose MTD of BPP-PTX was 100 mg/kg (equivalent to 48.7 μmol/kg). The weight loss was more severe as the drug dose increased. However, the acute weight loss caused by high dose was temporary, with the body weight recovering to baseline levels within 15 days. Then, we evaluated the plasma pharmacokinetics profiles of plain PTX and BPP-PTX. Mice xenografted with MDA-MB-468 received intraperitoneal (i.p.) injections of 15 mg/kg PTX (equivalent to 17.6 μmol/kg) or 36.1 mg/kg BPP-PTX (equivalent to 17.6 μmol/kg). The plasma PTX concentration in the plain PTX-treated group reached peak levels at 1 h and then decreased rapidly, and it was quickly removed from the circulating system. In contrast, the plasma PTX concentration in the BPP-PTX-treated group remained high for an extended period after 1 h, suggesting that BPP-PTX could have a lower release rate in circulation.
Click to Show/Hide
|
||||
| In Vivo Model | Female BALB/c mice. | ||||
| Half life period | 5.82 h | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [447] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Weight loss rate | 7.20% | |||
| Administration Dosage | 100 mg/kg | ||||
| MOA of PDC |
Here, we report the design, synthesis, and evaluation-in vitro and in vivo-of a novel PDC, namely BPP-PTX, whereby PTX is conjugated to one member of BPPs, Bj-BPP-9a (teprotide), via a succinyl linker. The targeting moiety was carefully selected based on previous studies. It was similarly employed with a nanoparticle carrier in vivo and was shown to modulate improved drug accumulation at the tumor site, thereby curbing tumor growth and extending the lives of tumor-bearing mice. In this study, we demonstrate for the first time that BPP-PTX functions through BPPs cognate receptor, ACE. ACE was overexpressed in TNBC cell lines but not in the receptor-positive cell line. BPP, as part of BPP-PTX, bound ACE and mediated its selective cytotoxic action through this receptor in ACE-positive TNBC cells. Furthermore, BPP-PTX demonstrated improved biodistribution, therapeutic activity, and better safety profile in vivo. These results advocate the significance of BPP-PTX as a suitable tumor-targeting PDC, strongly warranting further refinement and investigations for TNBC.
Click to Show/Hide
|
||||
| Description |
After obtaining positive in vitro results, BPP-PTX was then tested in vivo. To test the in vivo toxicity of the drug, the maximum tolerated dose (MTD) of BPP-PTX in Balb/c mice was determined. The single-dose MTD (acute toxicity) of plain PTX was 20 mg/kg (equivalent to 23.4 μmol/kg), similar to previous findings in the literature, while the single-dose MTD of BPP-PTX was 100 mg/kg (equivalent to 48.7 μmol/kg). The weight loss was more severe as the drug dose increased. However, the acute weight loss caused by high dose was temporary, with the body weight recovering to baseline levels within 15 days. Then, we evaluated the plasma pharmacokinetics profiles of plain PTX and BPP-PTX. Mice xenografted with MDA-MB-468 received intraperitoneal (i.p.) injections of 15 mg/kg PTX (equivalent to 17.6 μmol/kg) or 36.1 mg/kg BPP-PTX (equivalent to 17.6 μmol/kg). The plasma PTX concentration in the plain PTX-treated group reached peak levels at 1 h and then decreased rapidly, and it was quickly removed from the circulating system. In contrast, the plasma PTX concentration in the BPP-PTX-treated group remained high for an extended period after 1 h, suggesting that BPP-PTX could have a lower release rate in circulation.
Click to Show/Hide
|
||||
| In Vivo Model | Female BALB/c mice. | ||||
| Half life period | 5.82 h | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [447] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Mice survival rate | 75% | |||
| Administration Dosage | 120 mg/kg | ||||
| MOA of PDC |
Here, we report the design, synthesis, and evaluation-in vitro and in vivo-of a novel PDC, namely BPP-PTX, whereby PTX is conjugated to one member of BPPs, Bj-BPP-9a (teprotide), via a succinyl linker. The targeting moiety was carefully selected based on previous studies. It was similarly employed with a nanoparticle carrier in vivo and was shown to modulate improved drug accumulation at the tumor site, thereby curbing tumor growth and extending the lives of tumor-bearing mice. In this study, we demonstrate for the first time that BPP-PTX functions through BPPs cognate receptor, ACE. ACE was overexpressed in TNBC cell lines but not in the receptor-positive cell line. BPP, as part of BPP-PTX, bound ACE and mediated its selective cytotoxic action through this receptor in ACE-positive TNBC cells. Furthermore, BPP-PTX demonstrated improved biodistribution, therapeutic activity, and better safety profile in vivo. These results advocate the significance of BPP-PTX as a suitable tumor-targeting PDC, strongly warranting further refinement and investigations for TNBC.
Click to Show/Hide
|
||||
| Description |
After obtaining positive in vitro results, BPP-PTX was then tested in vivo. To test the in vivo toxicity of the drug, the maximum tolerated dose (MTD) of BPP-PTX in Balb/c mice was determined. The single-dose MTD (acute toxicity) of plain PTX was 20 mg/kg (equivalent to 23.4 μmol/kg), similar to previous findings in the literature, while the single-dose MTD of BPP-PTX was 100 mg/kg (equivalent to 48.7 μmol/kg). The weight loss was more severe as the drug dose increased. However, the acute weight loss caused by high dose was temporary, with the body weight recovering to baseline levels within 15 days. Then, we evaluated the plasma pharmacokinetics profiles of plain PTX and BPP-PTX. Mice xenografted with MDA-MB-468 received intraperitoneal (i.p.) injections of 15 mg/kg PTX (equivalent to 17.6 μmol/kg) or 36.1 mg/kg BPP-PTX (equivalent to 17.6 μmol/kg). The plasma PTX concentration in the plain PTX-treated group reached peak levels at 1 h and then decreased rapidly, and it was quickly removed from the circulating system. In contrast, the plasma PTX concentration in the BPP-PTX-treated group remained high for an extended period after 1 h, suggesting that BPP-PTX could have a lower release rate in circulation.
Click to Show/Hide
|
||||
| In Vivo Model | Female BALB/c mice. | ||||
| Half life period | 5.82 h | ||||
| Experiment 5 Reporting the Activity Data of This PDC | [447] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Mice survival rate | 100% | |||
| Administration Dosage | 60 mg/kg | ||||
| MOA of PDC |
Here, we report the design, synthesis, and evaluation-in vitro and in vivo-of a novel PDC, namely BPP-PTX, whereby PTX is conjugated to one member of BPPs, Bj-BPP-9a (teprotide), via a succinyl linker. The targeting moiety was carefully selected based on previous studies. It was similarly employed with a nanoparticle carrier in vivo and was shown to modulate improved drug accumulation at the tumor site, thereby curbing tumor growth and extending the lives of tumor-bearing mice. In this study, we demonstrate for the first time that BPP-PTX functions through BPPs cognate receptor, ACE. ACE was overexpressed in TNBC cell lines but not in the receptor-positive cell line. BPP, as part of BPP-PTX, bound ACE and mediated its selective cytotoxic action through this receptor in ACE-positive TNBC cells. Furthermore, BPP-PTX demonstrated improved biodistribution, therapeutic activity, and better safety profile in vivo. These results advocate the significance of BPP-PTX as a suitable tumor-targeting PDC, strongly warranting further refinement and investigations for TNBC.
Click to Show/Hide
|
||||
| Description |
After obtaining positive in vitro results, BPP-PTX was then tested in vivo. To test the in vivo toxicity of the drug, the maximum tolerated dose (MTD) of BPP-PTX in Balb/c mice was determined. The single-dose MTD (acute toxicity) of plain PTX was 20 mg/kg (equivalent to 23.4 μmol/kg), similar to previous findings in the literature, while the single-dose MTD of BPP-PTX was 100 mg/kg (equivalent to 48.7 μmol/kg). The weight loss was more severe as the drug dose increased. However, the acute weight loss caused by high dose was temporary, with the body weight recovering to baseline levels within 15 days. Then, we evaluated the plasma pharmacokinetics profiles of plain PTX and BPP-PTX. Mice xenografted with MDA-MB-468 received intraperitoneal (i.p.) injections of 15 mg/kg PTX (equivalent to 17.6 μmol/kg) or 36.1 mg/kg BPP-PTX (equivalent to 17.6 μmol/kg). The plasma PTX concentration in the plain PTX-treated group reached peak levels at 1 h and then decreased rapidly, and it was quickly removed from the circulating system. In contrast, the plasma PTX concentration in the BPP-PTX-treated group remained high for an extended period after 1 h, suggesting that BPP-PTX could have a lower release rate in circulation.
Click to Show/Hide
|
||||
| In Vivo Model | Female BALB/c mice. | ||||
| Half life period | 5.82 h | ||||
| Experiment 6 Reporting the Activity Data of This PDC | [447] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Mice survival rate | 100% | |||
| Administration Dosage | 80 mg/kg | ||||
| MOA of PDC |
Here, we report the design, synthesis, and evaluation-in vitro and in vivo-of a novel PDC, namely BPP-PTX, whereby PTX is conjugated to one member of BPPs, Bj-BPP-9a (teprotide), via a succinyl linker. The targeting moiety was carefully selected based on previous studies. It was similarly employed with a nanoparticle carrier in vivo and was shown to modulate improved drug accumulation at the tumor site, thereby curbing tumor growth and extending the lives of tumor-bearing mice. In this study, we demonstrate for the first time that BPP-PTX functions through BPPs cognate receptor, ACE. ACE was overexpressed in TNBC cell lines but not in the receptor-positive cell line. BPP, as part of BPP-PTX, bound ACE and mediated its selective cytotoxic action through this receptor in ACE-positive TNBC cells. Furthermore, BPP-PTX demonstrated improved biodistribution, therapeutic activity, and better safety profile in vivo. These results advocate the significance of BPP-PTX as a suitable tumor-targeting PDC, strongly warranting further refinement and investigations for TNBC.
Click to Show/Hide
|
||||
| Description |
After obtaining positive in vitro results, BPP-PTX was then tested in vivo. To test the in vivo toxicity of the drug, the maximum tolerated dose (MTD) of BPP-PTX in Balb/c mice was determined. The single-dose MTD (acute toxicity) of plain PTX was 20 mg/kg (equivalent to 23.4 μmol/kg), similar to previous findings in the literature, while the single-dose MTD of BPP-PTX was 100 mg/kg (equivalent to 48.7 μmol/kg). The weight loss was more severe as the drug dose increased. However, the acute weight loss caused by high dose was temporary, with the body weight recovering to baseline levels within 15 days. Then, we evaluated the plasma pharmacokinetics profiles of plain PTX and BPP-PTX. Mice xenografted with MDA-MB-468 received intraperitoneal (i.p.) injections of 15 mg/kg PTX (equivalent to 17.6 μmol/kg) or 36.1 mg/kg BPP-PTX (equivalent to 17.6 μmol/kg). The plasma PTX concentration in the plain PTX-treated group reached peak levels at 1 h and then decreased rapidly, and it was quickly removed from the circulating system. In contrast, the plasma PTX concentration in the BPP-PTX-treated group remained high for an extended period after 1 h, suggesting that BPP-PTX could have a lower release rate in circulation.
Click to Show/Hide
|
||||
| In Vivo Model | Female BALB/c mice. | ||||
| Half life period | 5.82 h | ||||
| Experiment 7 Reporting the Activity Data of This PDC | [447] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Mice survival rate | 100% | |||
| Administration Dosage | 100 mg/kg | ||||
| MOA of PDC |
Here, we report the design, synthesis, and evaluation-in vitro and in vivo-of a novel PDC, namely BPP-PTX, whereby PTX is conjugated to one member of BPPs, Bj-BPP-9a (teprotide), via a succinyl linker. The targeting moiety was carefully selected based on previous studies. It was similarly employed with a nanoparticle carrier in vivo and was shown to modulate improved drug accumulation at the tumor site, thereby curbing tumor growth and extending the lives of tumor-bearing mice. In this study, we demonstrate for the first time that BPP-PTX functions through BPPs cognate receptor, ACE. ACE was overexpressed in TNBC cell lines but not in the receptor-positive cell line. BPP, as part of BPP-PTX, bound ACE and mediated its selective cytotoxic action through this receptor in ACE-positive TNBC cells. Furthermore, BPP-PTX demonstrated improved biodistribution, therapeutic activity, and better safety profile in vivo. These results advocate the significance of BPP-PTX as a suitable tumor-targeting PDC, strongly warranting further refinement and investigations for TNBC.
Click to Show/Hide
|
||||
| Description |
After obtaining positive in vitro results, BPP-PTX was then tested in vivo. To test the in vivo toxicity of the drug, the maximum tolerated dose (MTD) of BPP-PTX in Balb/c mice was determined. The single-dose MTD (acute toxicity) of plain PTX was 20 mg/kg (equivalent to 23.4 μmol/kg), similar to previous findings in the literature, while the single-dose MTD of BPP-PTX was 100 mg/kg (equivalent to 48.7 μmol/kg). The weight loss was more severe as the drug dose increased. However, the acute weight loss caused by high dose was temporary, with the body weight recovering to baseline levels within 15 days. Then, we evaluated the plasma pharmacokinetics profiles of plain PTX and BPP-PTX. Mice xenografted with MDA-MB-468 received intraperitoneal (i.p.) injections of 15 mg/kg PTX (equivalent to 17.6 μmol/kg) or 36.1 mg/kg BPP-PTX (equivalent to 17.6 μmol/kg). The plasma PTX concentration in the plain PTX-treated group reached peak levels at 1 h and then decreased rapidly, and it was quickly removed from the circulating system. In contrast, the plasma PTX concentration in the BPP-PTX-treated group remained high for an extended period after 1 h, suggesting that BPP-PTX could have a lower release rate in circulation.
Click to Show/Hide
|
||||
| In Vivo Model | Female BALB/c mice. | ||||
| Half life period | 5.82 h | ||||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [447] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 9.5 nM | |||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Here, we report the design, synthesis, and evaluation-in vitro and in vivo-of a novel PDC, namely BPP-PTX, whereby PTX is conjugated to one member of BPPs, Bj-BPP-9a (teprotide), via a succinyl linker. The targeting moiety was carefully selected based on previous studies. It was similarly employed with a nanoparticle carrier in vivo and was shown to modulate improved drug accumulation at the tumor site, thereby curbing tumor growth and extending the lives of tumor-bearing mice. In this study, we demonstrate for the first time that BPP-PTX functions through BPPs cognate receptor, ACE. ACE was overexpressed in TNBC cell lines but not in the receptor-positive cell line. BPP, as part of BPP-PTX, bound ACE and mediated its selective cytotoxic action through this receptor in ACE-positive TNBC cells. Furthermore, BPP-PTX demonstrated improved biodistribution, therapeutic activity, and better safety profile in vivo. These results advocate the significance of BPP-PTX as a suitable tumor-targeting PDC, strongly warranting further refinement and investigations for TNBC.
Click to Show/Hide
|
||||
| Description |
To evaluate the in vitro antitumor activity, BPP-PTX, uncoupled peptide (BPP), and PTX were under cytotoxicity evaluation by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazole (MTT) assay in ACE-positive TNBC cell lines (MDA-MB-231 and MDA-MB-468) and ACE-negative cell lines (HEK293T). The free BPP peptide did not exhibit any cytotoxicity in all cell lines. In contrast, the IC50 value of BPP-PTX in ACE-negative HEK293T was 616.1 nM [95% CI, (242.7, 1564.0)], which was much higher than that of PTX in the same cell line {6.7 nM [95% CI, (5.3, 8.4)]}. Interestingly, the cytotoxicity of BPP-PTX was comparable with that of PTX in ACE-positive TNBC cell lines. In MDA-MB-231, the IC50 value of BPP-PTX was 9.5 nM [95% CI, (7.0, 12.8)], and that of PTX was 3.1 nM [95% CI, (2.8, 3.5)]. In MDA-MB-468, the IC50 value of BPP-PTX was 12.3 nM [95% CI, (6.8, 22.3)], and that of PTX was 3.0 nM [95% CI, (2.0, 4.7)].
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 (ACE+) cell | CVCL_0062 | ||
| Half life period | 5.82 h | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [447] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 12.3 nM | |||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Here, we report the design, synthesis, and evaluation-in vitro and in vivo-of a novel PDC, namely BPP-PTX, whereby PTX is conjugated to one member of BPPs, Bj-BPP-9a (teprotide), via a succinyl linker. The targeting moiety was carefully selected based on previous studies. It was similarly employed with a nanoparticle carrier in vivo and was shown to modulate improved drug accumulation at the tumor site, thereby curbing tumor growth and extending the lives of tumor-bearing mice. In this study, we demonstrate for the first time that BPP-PTX functions through BPPs cognate receptor, ACE. ACE was overexpressed in TNBC cell lines but not in the receptor-positive cell line. BPP, as part of BPP-PTX, bound ACE and mediated its selective cytotoxic action through this receptor in ACE-positive TNBC cells. Furthermore, BPP-PTX demonstrated improved biodistribution, therapeutic activity, and better safety profile in vivo. These results advocate the significance of BPP-PTX as a suitable tumor-targeting PDC, strongly warranting further refinement and investigations for TNBC.
Click to Show/Hide
|
||||
| Description |
To evaluate the in vitro antitumor activity, BPP-PTX, uncoupled peptide (BPP), and PTX were under cytotoxicity evaluation by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazole (MTT) assay in ACE-positive TNBC cell lines (MDA-MB-231 and MDA-MB-468) and ACE-negative cell lines (HEK293T). The free BPP peptide did not exhibit any cytotoxicity in all cell lines. In contrast, the IC50 value of BPP-PTX in ACE-negative HEK293T was 616.1 nM [95% CI, (242.7, 1564.0)], which was much higher than that of PTX in the same cell line {6.7 nM [95% CI, (5.3, 8.4)]}. Interestingly, the cytotoxicity of BPP-PTX was comparable with that of PTX in ACE-positive TNBC cell lines. In MDA-MB-231, the IC50 value of BPP-PTX was 9.5 nM [95% CI, (7.0, 12.8)], and that of PTX was 3.1 nM [95% CI, (2.8, 3.5)]. In MDA-MB-468, the IC50 value of BPP-PTX was 12.3 nM [95% CI, (6.8, 22.3)], and that of PTX was 3.0 nM [95% CI, (2.0, 4.7)].
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-468 cell | CVCL_0419 | ||
| Half life period | 5.82 h | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [447] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 616.1 nM | |||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Here, we report the design, synthesis, and evaluation-in vitro and in vivo-of a novel PDC, namely BPP-PTX, whereby PTX is conjugated to one member of BPPs, Bj-BPP-9a (teprotide), via a succinyl linker. The targeting moiety was carefully selected based on previous studies. It was similarly employed with a nanoparticle carrier in vivo and was shown to modulate improved drug accumulation at the tumor site, thereby curbing tumor growth and extending the lives of tumor-bearing mice. In this study, we demonstrate for the first time that BPP-PTX functions through BPPs cognate receptor, ACE. ACE was overexpressed in TNBC cell lines but not in the receptor-positive cell line. BPP, as part of BPP-PTX, bound ACE and mediated its selective cytotoxic action through this receptor in ACE-positive TNBC cells. Furthermore, BPP-PTX demonstrated improved biodistribution, therapeutic activity, and better safety profile in vivo. These results advocate the significance of BPP-PTX as a suitable tumor-targeting PDC, strongly warranting further refinement and investigations for TNBC.
Click to Show/Hide
|
||||
| Description |
To evaluate the in vitro antitumor activity, BPP-PTX, uncoupled peptide (BPP), and PTX were under cytotoxicity evaluation by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazole (MTT) assay in ACE-positive TNBC cell lines (MDA-MB-231 and MDA-MB-468) and ACE-negative cell lines (HEK293T). The free BPP peptide did not exhibit any cytotoxicity in all cell lines. In contrast, the IC50 value of BPP-PTX in ACE-negative HEK293T was 616.1 nM [95% CI, (242.7, 1564.0)], which was much higher than that of PTX in the same cell line {6.7 nM [95% CI, (5.3, 8.4)]}. Interestingly, the cytotoxicity of BPP-PTX was comparable with that of PTX in ACE-positive TNBC cell lines. In MDA-MB-231, the IC50 value of BPP-PTX was 9.5 nM [95% CI, (7.0, 12.8)], and that of PTX was 3.1 nM [95% CI, (2.8, 3.5)]. In MDA-MB-468, the IC50 value of BPP-PTX was 12.3 nM [95% CI, (6.8, 22.3)], and that of PTX was 3.0 nM [95% CI, (2.0, 4.7)].
Click to Show/Hide
|
||||
| In Vitro Model | Normal | HEK-293T (ACE-) cell | CVCL_0063 | ||
| Half life period | 5.82 h | ||||
LTP-1 [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [448] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 67.10% | |||
| Administration Time | Every two days for 2 weeks | ||||
| Administration Dosage | 8 μmol/kg | ||||
| MOA of PDC |
LHRH, also named gonadotropin-releasing hormone (GnRH), is an endogenous peptide agonist (primary sequence: pGlu-His-Trp-Ser-Tyr-Gly-Leu-Arg-Pro-Gly-NH2) released from hypothalamus. LHRH-R (LHRH receptor), a member of the G protein-coupled receptor family, is overexpressed in various tumor types, while their expression in the corresponding normal tissues, apart from pituitary cells, is comparatively low. Given this, we chose LHRH as the TTP component of MSCPTP (TTP-CPP peptide). In this investigation, we combined LHRH (as the TTP part), peptide PLGLAG, T2 (as the CPP part) and cysteine (as linker binding site) into an MSCPTP named LT-1. Then PTX was conjugated with LT-1 via a GSH-cleavable module to produce the smart PDC, namely LTP-1 (TTP-CPP-PTX conjugate). In vitro, LTP-1 exhibited selective and stronger cytotoxicity than PTX against LHRH-R-positive tumor cells with little effect on normal cells. In vivo, LTP-1 was highly effective in suppressing tumor growth in an MCF-7 xenograft mouse model. Additional experiments on both cellular and molecular levels were carried out to unravel the possible antitumor mechanism of action of LTP-1.
Click to Show/Hide
|
||||
| Description |
At the dose of 8 μmol/kg, LTP-1 decreased the tumor volume and tumor weight by 77.2% and 67.1%.
|
||||
| In Vivo Model | MCF-7 xenograft mice. | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [448] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 77.20% | |||
| Evaluation Method | Tumor volume detection | ||||
| Administration Time | Every two days for 2 weeks | ||||
| Administration Dosage | 8 μmol/kg | ||||
| MOA of PDC |
LHRH, also named gonadotropin-releasing hormone (GnRH), is an endogenous peptide agonist (primary sequence: pGlu-His-Trp-Ser-Tyr-Gly-Leu-Arg-Pro-Gly-NH2) released from hypothalamus. LHRH-R (LHRH receptor), a member of the G protein-coupled receptor family, is overexpressed in various tumor types, while their expression in the corresponding normal tissues, apart from pituitary cells, is comparatively low. Given this, we chose LHRH as the TTP component of MSCPTP (TTP-CPP peptide). In this investigation, we combined LHRH (as the TTP part), peptide PLGLAG, T2 (as the CPP part) and cysteine (as linker binding site) into an MSCPTP named LT-1. Then PTX was conjugated with LT-1 via a GSH-cleavable module to produce the smart PDC, namely LTP-1 (TTP-CPP-PTX conjugate). In vitro, LTP-1 exhibited selective and stronger cytotoxicity than PTX against LHRH-R-positive tumor cells with little effect on normal cells. In vivo, LTP-1 was highly effective in suppressing tumor growth in an MCF-7 xenograft mouse model. Additional experiments on both cellular and molecular levels were carried out to unravel the possible antitumor mechanism of action of LTP-1.
Click to Show/Hide
|
||||
| Description |
At the dose of 8 μmol/kg, LTP-1 decreased the tumor volume and tumor weight by 77.2% and 67.1%.
|
||||
| In Vivo Model | MCF-7 xenograft mice. | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [448] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 83.40% | |||
| Administration Time | Every two days for 2 weeks | ||||
| Administration Dosage | 12 μmol/kg | ||||
| MOA of PDC |
LHRH, also named gonadotropin-releasing hormone (GnRH), is an endogenous peptide agonist (primary sequence: pGlu-His-Trp-Ser-Tyr-Gly-Leu-Arg-Pro-Gly-NH2) released from hypothalamus. LHRH-R (LHRH receptor), a member of the G protein-coupled receptor family, is overexpressed in various tumor types, while their expression in the corresponding normal tissues, apart from pituitary cells, is comparatively low. Given this, we chose LHRH as the TTP component of MSCPTP (TTP-CPP peptide). In this investigation, we combined LHRH (as the TTP part), peptide PLGLAG, T2 (as the CPP part) and cysteine (as linker binding site) into an MSCPTP named LT-1. Then PTX was conjugated with LT-1 via a GSH-cleavable module to produce the smart PDC, namely LTP-1 (TTP-CPP-PTX conjugate). In vitro, LTP-1 exhibited selective and stronger cytotoxicity than PTX against LHRH-R-positive tumor cells with little effect on normal cells. In vivo, LTP-1 was highly effective in suppressing tumor growth in an MCF-7 xenograft mouse model. Additional experiments on both cellular and molecular levels were carried out to unravel the possible antitumor mechanism of action of LTP-1.
Click to Show/Hide
|
||||
| Description |
At the dose of 12 μmol/kg, LTP-1 decreased the tumor volume and tumor weight by 90.1% and 83.4%, respectively.
|
||||
| In Vivo Model | MCF-7 xenograft mice. | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [448] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 90.10% | |||
| Evaluation Method | Tumor volume detection | ||||
| Administration Time | Every two days for 2 weeks | ||||
| Administration Dosage | 12 μmol/kg | ||||
| MOA of PDC |
LHRH, also named gonadotropin-releasing hormone (GnRH), is an endogenous peptide agonist (primary sequence: pGlu-His-Trp-Ser-Tyr-Gly-Leu-Arg-Pro-Gly-NH2) released from hypothalamus. LHRH-R (LHRH receptor), a member of the G protein-coupled receptor family, is overexpressed in various tumor types, while their expression in the corresponding normal tissues, apart from pituitary cells, is comparatively low. Given this, we chose LHRH as the TTP component of MSCPTP (TTP-CPP peptide). In this investigation, we combined LHRH (as the TTP part), peptide PLGLAG, T2 (as the CPP part) and cysteine (as linker binding site) into an MSCPTP named LT-1. Then PTX was conjugated with LT-1 via a GSH-cleavable module to produce the smart PDC, namely LTP-1 (TTP-CPP-PTX conjugate). In vitro, LTP-1 exhibited selective and stronger cytotoxicity than PTX against LHRH-R-positive tumor cells with little effect on normal cells. In vivo, LTP-1 was highly effective in suppressing tumor growth in an MCF-7 xenograft mouse model. Additional experiments on both cellular and molecular levels were carried out to unravel the possible antitumor mechanism of action of LTP-1.
Click to Show/Hide
|
||||
| Description |
At the dose of 12 μmol/kg, LTP-1 decreased the tumor volume and tumor weight by 90.1% and 83.4%, respectively.
|
||||
| In Vivo Model | MCF-7 xenograft mice. | ||||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [448] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 3.8 ± 0.3 nM | |||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
LHRH, also named gonadotropin-releasing hormone (GnRH), is an endogenous peptide agonist (primary sequence: pGlu-His-Trp-Ser-Tyr-Gly-Leu-Arg-Pro-Gly-NH2) released from hypothalamus. LHRH-R (LHRH receptor), a member of the G protein-coupled receptor family, is overexpressed in various tumor types, while their expression in the corresponding normal tissues, apart from pituitary cells, is comparatively low. Given this, we chose LHRH as the TTP component of MSCPTP (TTP-CPP peptide). In this investigation, we combined LHRH (as the TTP part), peptide PLGLAG, T2 (as the CPP part) and cysteine (as linker binding site) into an MSCPTP named LT-1. Then PTX was conjugated with LT-1 via a GSH-cleavable module to produce the smart PDC, namely LTP-1 (TTP-CPP-PTX conjugate). In vitro, LTP-1 exhibited selective and stronger cytotoxicity than PTX against LHRH-R-positive tumor cells with little effect on normal cells. In vivo, LTP-1 was highly effective in suppressing tumor growth in an MCF-7 xenograft mouse model. Additional experiments on both cellular and molecular levels were carried out to unravel the possible antitumor mechanism of action of LTP-1.
Click to Show/Hide
|
||||
| Description |
LTP-1 exhibited greater anti-proliferative effects (IC50s of 3.8-20.3 nM) than PTX (IC50s of 6.6-28.6 nM) against most cancer cells except Hela, and less cytotoxicity to normal cells (IC50s of >80 nM and 66.0 nM for NCM460 and HEK-293, respectively). Thus, LTP-1 displayed not only enhanced anti-proliferative activity, but also higher selectivity for cancer cells over normal cells. It is also worthy of note that LTP-1 showed much higher activity against the paclitaxel-resistant A2780/PTX cells with an IC50 of 0.8 μM, as compared to PTX which is essentially inactive (IC50 = 23.9 μM). Hemolysis assay further testified that LTP-1 presented weak hemolytic activity even at a concentration up to 80 μM (as illustrated in Fig. 4a).
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [448] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 5.6 ± 0.2 nM | |||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
LHRH, also named gonadotropin-releasing hormone (GnRH), is an endogenous peptide agonist (primary sequence: pGlu-His-Trp-Ser-Tyr-Gly-Leu-Arg-Pro-Gly-NH2) released from hypothalamus. LHRH-R (LHRH receptor), a member of the G protein-coupled receptor family, is overexpressed in various tumor types, while their expression in the corresponding normal tissues, apart from pituitary cells, is comparatively low. Given this, we chose LHRH as the TTP component of MSCPTP (TTP-CPP peptide). In this investigation, we combined LHRH (as the TTP part), peptide PLGLAG, T2 (as the CPP part) and cysteine (as linker binding site) into an MSCPTP named LT-1. Then PTX was conjugated with LT-1 via a GSH-cleavable module to produce the smart PDC, namely LTP-1 (TTP-CPP-PTX conjugate). In vitro, LTP-1 exhibited selective and stronger cytotoxicity than PTX against LHRH-R-positive tumor cells with little effect on normal cells. In vivo, LTP-1 was highly effective in suppressing tumor growth in an MCF-7 xenograft mouse model. Additional experiments on both cellular and molecular levels were carried out to unravel the possible antitumor mechanism of action of LTP-1.
Click to Show/Hide
|
||||
| Description |
LTP-1 exhibited greater anti-proliferative effects (IC50s of 3.8-20.3 nM) than PTX (IC50s of 6.6-28.6 nM) against most cancer cells except Hela, and less cytotoxicity to normal cells (IC50s of >80 nM and 66.0 nM for NCM460 and HEK-293, respectively). Thus, LTP-1 displayed not only enhanced anti-proliferative activity, but also higher selectivity for cancer cells over normal cells. It is also worthy of note that LTP-1 showed much higher activity against the paclitaxel-resistant A2780/PTX cells with an IC50 of 0.8 μM, as compared to PTX which is essentially inactive (IC50 = 23.9 μM). Hemolysis assay further testified that LTP-1 presented weak hemolytic activity even at a concentration up to 80 μM (as illustrated in Fig. 4a).
Click to Show/Hide
|
||||
| In Vitro Model | Colon adenocarcinoma | HT-29 cell | CVCL_0320 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [448] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 8.3 ± 0.5 nM | |||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
LHRH, also named gonadotropin-releasing hormone (GnRH), is an endogenous peptide agonist (primary sequence: pGlu-His-Trp-Ser-Tyr-Gly-Leu-Arg-Pro-Gly-NH2) released from hypothalamus. LHRH-R (LHRH receptor), a member of the G protein-coupled receptor family, is overexpressed in various tumor types, while their expression in the corresponding normal tissues, apart from pituitary cells, is comparatively low. Given this, we chose LHRH as the TTP component of MSCPTP (TTP-CPP peptide). In this investigation, we combined LHRH (as the TTP part), peptide PLGLAG, T2 (as the CPP part) and cysteine (as linker binding site) into an MSCPTP named LT-1. Then PTX was conjugated with LT-1 via a GSH-cleavable module to produce the smart PDC, namely LTP-1 (TTP-CPP-PTX conjugate). In vitro, LTP-1 exhibited selective and stronger cytotoxicity than PTX against LHRH-R-positive tumor cells with little effect on normal cells. In vivo, LTP-1 was highly effective in suppressing tumor growth in an MCF-7 xenograft mouse model. Additional experiments on both cellular and molecular levels were carried out to unravel the possible antitumor mechanism of action of LTP-1.
Click to Show/Hide
|
||||
| Description |
LTP-1 exhibited greater anti-proliferative effects (IC50s of 3.8-20.3 nM) than PTX (IC50s of 6.6-28.6 nM) against most cancer cells except Hela, and less cytotoxicity to normal cells (IC50s of >80 nM and 66.0 nM for NCM460 and HEK-293, respectively). Thus, LTP-1 displayed not only enhanced anti-proliferative activity, but also higher selectivity for cancer cells over normal cells. It is also worthy of note that LTP-1 showed much higher activity against the paclitaxel-resistant A2780/PTX cells with an IC50 of 0.8 μM, as compared to PTX which is essentially inactive (IC50 = 23.9 μM). Hemolysis assay further testified that LTP-1 presented weak hemolytic activity even at a concentration up to 80 μM (as illustrated in Fig. 4a).
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [448] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 20.3 ± 3.3 nM | |||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
LHRH, also named gonadotropin-releasing hormone (GnRH), is an endogenous peptide agonist (primary sequence: pGlu-His-Trp-Ser-Tyr-Gly-Leu-Arg-Pro-Gly-NH2) released from hypothalamus. LHRH-R (LHRH receptor), a member of the G protein-coupled receptor family, is overexpressed in various tumor types, while their expression in the corresponding normal tissues, apart from pituitary cells, is comparatively low. Given this, we chose LHRH as the TTP component of MSCPTP (TTP-CPP peptide). In this investigation, we combined LHRH (as the TTP part), peptide PLGLAG, T2 (as the CPP part) and cysteine (as linker binding site) into an MSCPTP named LT-1. Then PTX was conjugated with LT-1 via a GSH-cleavable module to produce the smart PDC, namely LTP-1 (TTP-CPP-PTX conjugate). In vitro, LTP-1 exhibited selective and stronger cytotoxicity than PTX against LHRH-R-positive tumor cells with little effect on normal cells. In vivo, LTP-1 was highly effective in suppressing tumor growth in an MCF-7 xenograft mouse model. Additional experiments on both cellular and molecular levels were carried out to unravel the possible antitumor mechanism of action of LTP-1.
Click to Show/Hide
|
||||
| Description |
LTP-1 exhibited greater anti-proliferative effects (IC50s of 3.8-20.3 nM) than PTX (IC50s of 6.6-28.6 nM) against most cancer cells except Hela, and less cytotoxicity to normal cells (IC50s of >80 nM and 66.0 nM for NCM460 and HEK-293, respectively). Thus, LTP-1 displayed not only enhanced anti-proliferative activity, but also higher selectivity for cancer cells over normal cells. It is also worthy of note that LTP-1 showed much higher activity against the paclitaxel-resistant A2780/PTX cells with an IC50 of 0.8 μM, as compared to PTX which is essentially inactive (IC50 = 23.9 μM). Hemolysis assay further testified that LTP-1 presented weak hemolytic activity even at a concentration up to 80 μM (as illustrated in Fig. 4a).
Click to Show/Hide
|
||||
| In Vitro Model | Human papillomavirus-related cervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [448] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 66.0 ± 8.3 nM | |||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
LHRH, also named gonadotropin-releasing hormone (GnRH), is an endogenous peptide agonist (primary sequence: pGlu-His-Trp-Ser-Tyr-Gly-Leu-Arg-Pro-Gly-NH2) released from hypothalamus. LHRH-R (LHRH receptor), a member of the G protein-coupled receptor family, is overexpressed in various tumor types, while their expression in the corresponding normal tissues, apart from pituitary cells, is comparatively low. Given this, we chose LHRH as the TTP component of MSCPTP (TTP-CPP peptide). In this investigation, we combined LHRH (as the TTP part), peptide PLGLAG, T2 (as the CPP part) and cysteine (as linker binding site) into an MSCPTP named LT-1. Then PTX was conjugated with LT-1 via a GSH-cleavable module to produce the smart PDC, namely LTP-1 (TTP-CPP-PTX conjugate). In vitro, LTP-1 exhibited selective and stronger cytotoxicity than PTX against LHRH-R-positive tumor cells with little effect on normal cells. In vivo, LTP-1 was highly effective in suppressing tumor growth in an MCF-7 xenograft mouse model. Additional experiments on both cellular and molecular levels were carried out to unravel the possible antitumor mechanism of action of LTP-1.
Click to Show/Hide
|
||||
| Description |
LTP-1 exhibited greater anti-proliferative effects (IC50s of 3.8-20.3 nM) than PTX (IC50s of 6.6-28.6 nM) against most cancer cells except Hela, and less cytotoxicity to normal cells (IC50s of >80 nM and 66.0 nM for NCM460 and HEK-293, respectively). Thus, LTP-1 displayed not only enhanced anti-proliferative activity, but also higher selectivity for cancer cells over normal cells. It is also worthy of note that LTP-1 showed much higher activity against the paclitaxel-resistant A2780/PTX cells with an IC50 of 0.8 μM, as compared to PTX which is essentially inactive (IC50 = 23.9 μM). Hemolysis assay further testified that LTP-1 presented weak hemolytic activity even at a concentration up to 80 μM (as illustrated in Fig. 4a).
Click to Show/Hide
|
||||
| In Vitro Model | Normal | HEK293 cell | CVCL_0045 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [448] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 80.0 nM | |||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
LHRH, also named gonadotropin-releasing hormone (GnRH), is an endogenous peptide agonist (primary sequence: pGlu-His-Trp-Ser-Tyr-Gly-Leu-Arg-Pro-Gly-NH2) released from hypothalamus. LHRH-R (LHRH receptor), a member of the G protein-coupled receptor family, is overexpressed in various tumor types, while their expression in the corresponding normal tissues, apart from pituitary cells, is comparatively low. Given this, we chose LHRH as the TTP component of MSCPTP (TTP-CPP peptide). In this investigation, we combined LHRH (as the TTP part), peptide PLGLAG, T2 (as the CPP part) and cysteine (as linker binding site) into an MSCPTP named LT-1. Then PTX was conjugated with LT-1 via a GSH-cleavable module to produce the smart PDC, namely LTP-1 (TTP-CPP-PTX conjugate). In vitro, LTP-1 exhibited selective and stronger cytotoxicity than PTX against LHRH-R-positive tumor cells with little effect on normal cells. In vivo, LTP-1 was highly effective in suppressing tumor growth in an MCF-7 xenograft mouse model. Additional experiments on both cellular and molecular levels were carried out to unravel the possible antitumor mechanism of action of LTP-1.
Click to Show/Hide
|
||||
| Description |
LTP-1 exhibited greater anti-proliferative effects (IC50s of 3.8-20.3 nM) than PTX (IC50s of 6.6-28.6 nM) against most cancer cells except Hela, and less cytotoxicity to normal cells (IC50s of >80 nM and 66.0 nM for NCM460 and HEK-293, respectively). Thus, LTP-1 displayed not only enhanced anti-proliferative activity, but also higher selectivity for cancer cells over normal cells. It is also worthy of note that LTP-1 showed much higher activity against the paclitaxel-resistant A2780/PTX cells with an IC50 of 0.8 μM, as compared to PTX which is essentially inactive (IC50 = 23.9 μM). Hemolysis assay further testified that LTP-1 presented weak hemolytic activity even at a concentration up to 80 μM (as illustrated in Fig. 4a).
Click to Show/Hide
|
||||
| In Vitro Model | Normal | NCM460 cell | CVCL_0460 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [448] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 800 ± 100 nM | |||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
LHRH, also named gonadotropin-releasing hormone (GnRH), is an endogenous peptide agonist (primary sequence: pGlu-His-Trp-Ser-Tyr-Gly-Leu-Arg-Pro-Gly-NH2) released from hypothalamus. LHRH-R (LHRH receptor), a member of the G protein-coupled receptor family, is overexpressed in various tumor types, while their expression in the corresponding normal tissues, apart from pituitary cells, is comparatively low. Given this, we chose LHRH as the TTP component of MSCPTP (TTP-CPP peptide). In this investigation, we combined LHRH (as the TTP part), peptide PLGLAG, T2 (as the CPP part) and cysteine (as linker binding site) into an MSCPTP named LT-1. Then PTX was conjugated with LT-1 via a GSH-cleavable module to produce the smart PDC, namely LTP-1 (TTP-CPP-PTX conjugate). In vitro, LTP-1 exhibited selective and stronger cytotoxicity than PTX against LHRH-R-positive tumor cells with little effect on normal cells. In vivo, LTP-1 was highly effective in suppressing tumor growth in an MCF-7 xenograft mouse model. Additional experiments on both cellular and molecular levels were carried out to unravel the possible antitumor mechanism of action of LTP-1.
Click to Show/Hide
|
||||
| Description |
LTP-1 exhibited greater anti-proliferative effects (IC50s of 3.8-20.3 nM) than PTX (IC50s of 6.6-28.6 nM) against most cancer cells except Hela, and less cytotoxicity to normal cells (IC50s of >80 nM and 66.0 nM for NCM460 and HEK-293, respectively). Thus, LTP-1 displayed not only enhanced anti-proliferative activity, but also higher selectivity for cancer cells over normal cells. It is also worthy of note that LTP-1 showed much higher activity against the paclitaxel-resistant A2780/PTX cells with an IC50 of 0.8 μM, as compared to PTX which is essentially inactive (IC50 = 23.9 μM). Hemolysis assay further testified that LTP-1 presented weak hemolytic activity even at a concentration up to 80 μM (as illustrated in Fig. 4a).
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780/PTX cell | CVCL_C0D6 | ||
M1-PTX [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Glioma | ||||
| Efficacy Data | Percent survival | 0% | |||
| Administration Time | 25 days | ||||
| Administration Dosage | Normalized to 17.3 mg/kg PTX | ||||
| Description |
The survival of the PTX group was markedly lower than that of the Vehicle group. M1-PTX did not improve survival, whereas the survival of the M1-RGD-PTX group was increased.
|
||||
| In Vivo Model | U87MG-Luc-bearing xenograft model. | ||||
| In Vitro Model | Glioblastoma | U87MG-Luc cell | CVCL_5J15 | ||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Glioma | ||||
| Efficacy Data | Cell viability | 60% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 24 h | ||||
| Administration Dosage | 100 nM | ||||
| Description |
Both PDCs showed higher in vitro cytotoxicity than free PTX.
|
||||
| In Vitro Model | Glioblastoma | U-87MG cell | CVCL_0022 | ||
M1-RGD-PTX [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Glioma | ||||
| Efficacy Data | Percent survival | 20% | |||
| Administration Time | 25 days | ||||
| Administration Dosage | Normalized to 17.3 mg/kg PTX | ||||
| Description |
The survival of the PTX group was markedly lower than that of the Vehicle group. M1-PTX did not improve survival, whereas the survival of the M1-RGD-PTX group was increased.
|
||||
| In Vivo Model | U87MG-Luc-bearing xenograft model. | ||||
| In Vitro Model | Glioblastoma | U87MG-Luc cell | CVCL_5J15 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Percent survival | 50% | |||
| Administration Time | 20 days | ||||
| Administration Dosage | 44 mg/kg | ||||
| Description |
PTX did not increase survival, while the PDC M1-RGD-PTX markedly increased survival.
|
||||
| In Vivo Model | MDA-MB-231BR mouse model. | ||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Glioma | ||||
| Efficacy Data | Median lethal dose (LD50) | 152 mg/kg | |||
| Administration Time | 24 h | ||||
| Administration Dosage | 100 nM | ||||
| Description |
The fitted LD50 for M1-RGD-PTX was 152 mg/kg (equivalent to 60 mg/kg PTX).
|
||||
| In Vitro Model | Glioblastoma | U-87MG cell | CVCL_0022 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Glioma | ||||
| Efficacy Data | Cell viability | 55% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 24 h | ||||
| Administration Dosage | 100 nM | ||||
| Description |
Both PDCs showed higher in vitro cytotoxicity than free PTX.
|
||||
| In Vitro Model | Glioblastoma | U-87MG cell | CVCL_0022 | ||
(123B9)2L2PTX [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [449] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Number of Lung metastases nodules | 5 | |||
| Administration Time | 2 weeks | ||||
| Administration Dosage | 24.5 mg/kg | ||||
| Description |
We observed the clear and significant (p < 0.0001) beneficial effects of the dimer drug on lung metastasis (Figure5A,B), with a reduction of the gross lung-metastasis count by more than 75% compared with those in the control and Abraxane groups.
|
||||
| In Vivo Model | BALB/c mice syngeneic breast-cancer metastasis model. | ||||
SynB3PVGLIGPTX [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [450] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 68.20% | |||
| Evaluation Method | Bioluminescent imaging method | ||||
| Administration Time | 14 days | ||||
| Administration Dosage | 15 mg/5 mL kg-1 | ||||
| MOA of PDC |
SynB3PVGLIGPTX, was designed, screened, and synthetized, in which PTX was combined with CPPs (e.g., SynB3) through an MMP2sensitive linker (PVGLIG). This peptidedrug complex exhibits three advantages as follows: 1) the constructed structure of CPPs should help enhance the permeability of PTXcontaining nanocomplex across the BBB; 2) this nanocomplex covers a MMP2sensitive linker between CPPs and PTX, making it possible to release the drug at the target site with high MMP2 expression level; and 3) the novel dualfunctional PTX prodrug is a watersoluble nanocomplex, which can overcome the side effect exerted by low solubility and formulating agent (Cremophore EL) of PTX.
Click to Show/Hide
|
||||
| Description |
While after the administration of SynB3PVGLIGPTX, there were few differences in the tumor signals between day 7 and day 14 (Figure 6A). At all later days, the differences in log (BLI) between the SynB3PVGLIGPTX and control group were significantly increased (***p < 0.001, Figure 6B), in which the mouse treated with SynB3PVGLIGPTX yielded a notably weaker bioluminescence radiance and a concurrently smaller bioluminescent area than those in the control group. On day 28, mice treated with SynB3PVGLIGPTX achieved the highest tumor inhibition rate (91.40±0.57%), which was 2.27fold and 1.30fold higher than that of the PTX and TMZ groups, respectively (***p < 0.001, Figure 6C). From the results of the bioluminescence assays, we concluded that SynB3PVGLIGPTX could sufficiently suppress the growth of intracerebral tumors in vivo after 28 days compared with the inhibition rate yielded by TMZ or PTX (***p < 0.001, Figure 6C). In addition, the weight and overall survival period of mice administered with SynB3PVGLIGPTX (15 mg/5 mL kg-1, i.v.) were significantly higher than those in the control group (*p < 0.05, **p < 0.01, ***p < 0.001, Figure 6D,,E).E). Again, the separation in survival curves was notable among the four groups, with survival medians equal to 23, 29, and 25 for the control group, TMZ group and PTX group, respectively. In contrast, there was no death in the SynB3PVGLIGPTX group across the assays. Especially concerning was that, the administration concentration of SynB3PVGLIGPTX was 15 mg/5 mL kg-1, which is equivalent to 5.25 μmol/5 mL kg-1. Theoretically, after SynB3PVGLIGPTX at this concentration is completely hydrolyzed by MMP2, the concentration of free PTX released is 4.49 mg/5 mL kg-1, which is only onethird of the concentration administered in the PTX group. The experimental results show that, compared with the group given a high dose of PTX (15 mg/5 mL kg-1), the tumor volume of mice given the nanoconjugate (SynB3PVGLIGPTX) was significantly reduced, and its overall survival was also significantly prolonged, which indicates that SynB3PVGLIGPTX has a significant antiglioma activity in vivo. Accordingly, SynB3PVGLIGPTX, as a PDC that combined PTX to a dualfunctional peptide consisting of SynB3 and a MMP2sensitive peptide, could observably improve survival and decrease weight loss over the PTX monotherapy in vivo, and significantly overmatched the mice treated with TMZ.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c athymic nude mice xenograft model. | ||||
| In Vitro Model | Glioblastoma | U-87MG cell | CVCL_0022 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [450] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 70.32% | |||
| Evaluation Method | Bioluminescent imaging method | ||||
| Administration Time | 21 days | ||||
| Administration Dosage | 15 mg/5 mL kg-1 | ||||
| MOA of PDC |
SynB3PVGLIGPTX, was designed, screened, and synthetized, in which PTX was combined with CPPs (e.g., SynB3) through an MMP2sensitive linker (PVGLIG). This peptidedrug complex exhibits three advantages as follows: 1) the constructed structure of CPPs should help enhance the permeability of PTXcontaining nanocomplex across the BBB; 2) this nanocomplex covers a MMP2sensitive linker between CPPs and PTX, making it possible to release the drug at the target site with high MMP2 expression level; and 3) the novel dualfunctional PTX prodrug is a watersoluble nanocomplex, which can overcome the side effect exerted by low solubility and formulating agent (Cremophore EL) of PTX.
Click to Show/Hide
|
||||
| Description |
While after the administration of SynB3PVGLIGPTX, there were few differences in the tumor signals between day 7 and day 14 (Figure 6A). At all later days, the differences in log (BLI) between the SynB3PVGLIGPTX and control group were significantly increased (***p < 0.001, Figure 6B), in which the mouse treated with SynB3PVGLIGPTX yielded a notably weaker bioluminescence radiance and a concurrently smaller bioluminescent area than those in the control group. On day 28, mice treated with SynB3PVGLIGPTX achieved the highest tumor inhibition rate (91.40±0.57%), which was 2.27fold and 1.30fold higher than that of the PTX and TMZ groups, respectively (***p < 0.001, Figure 6C). From the results of the bioluminescence assays, we concluded that SynB3PVGLIGPTX could sufficiently suppress the growth of intracerebral tumors in vivo after 28 days compared with the inhibition rate yielded by TMZ or PTX (***p < 0.001, Figure 6C). In addition, the weight and overall survival period of mice administered with SynB3PVGLIGPTX (15 mg/5 mL kg-1, i.v.) were significantly higher than those in the control group (*p < 0.05, **p < 0.01, ***p < 0.001, Figure 6D,,E).E). Again, the separation in survival curves was notable among the four groups, with survival medians equal to 23, 29, and 25 for the control group, TMZ group and PTX group, respectively. In contrast, there was no death in the SynB3PVGLIGPTX group across the assays. Especially concerning was that, the administration concentration of SynB3PVGLIGPTX was 15 mg/5 mL kg-1, which is equivalent to 5.25 μmol/5 mL kg-1. Theoretically, after SynB3PVGLIGPTX at this concentration is completely hydrolyzed by MMP2, the concentration of free PTX released is 4.49 mg/5 mL kg-1, which is only onethird of the concentration administered in the PTX group. The experimental results show that, compared with the group given a high dose of PTX (15 mg/5 mL kg-1), the tumor volume of mice given the nanoconjugate (SynB3PVGLIGPTX) was significantly reduced, and its overall survival was also significantly prolonged, which indicates that SynB3PVGLIGPTX has a significant antiglioma activity in vivo. Accordingly, SynB3PVGLIGPTX, as a PDC that combined PTX to a dualfunctional peptide consisting of SynB3 and a MMP2sensitive peptide, could observably improve survival and decrease weight loss over the PTX monotherapy in vivo, and significantly overmatched the mice treated with TMZ.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c athymic nude mice xenograft model. | ||||
| In Vitro Model | Glioblastoma | U-87MG cell | CVCL_0022 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [450] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 91.50% | |||
| Evaluation Method | Bioluminescent imaging method | ||||
| Administration Time | 28 days | ||||
| Administration Dosage | 15 mg/5 mL kg-1 | ||||
| MOA of PDC |
SynB3PVGLIGPTX, was designed, screened, and synthetized, in which PTX was combined with CPPs (e.g., SynB3) through an MMP2sensitive linker (PVGLIG). This peptidedrug complex exhibits three advantages as follows: 1) the constructed structure of CPPs should help enhance the permeability of PTXcontaining nanocomplex across the BBB; 2) this nanocomplex covers a MMP2sensitive linker between CPPs and PTX, making it possible to release the drug at the target site with high MMP2 expression level; and 3) the novel dualfunctional PTX prodrug is a watersoluble nanocomplex, which can overcome the side effect exerted by low solubility and formulating agent (Cremophore EL) of PTX.
Click to Show/Hide
|
||||
| Description |
While after the administration of SynB3PVGLIGPTX, there were few differences in the tumor signals between day 7 and day 14 (Figure 6A). At all later days, the differences in log (BLI) between the SynB3PVGLIGPTX and control group were significantly increased (***p < 0.001, Figure 6B), in which the mouse treated with SynB3PVGLIGPTX yielded a notably weaker bioluminescence radiance and a concurrently smaller bioluminescent area than those in the control group. On day 28, mice treated with SynB3PVGLIGPTX achieved the highest tumor inhibition rate (91.40±0.57%), which was 2.27fold and 1.30fold higher than that of the PTX and TMZ groups, respectively (***p < 0.001, Figure 6C). From the results of the bioluminescence assays, we concluded that SynB3PVGLIGPTX could sufficiently suppress the growth of intracerebral tumors in vivo after 28 days compared with the inhibition rate yielded by TMZ or PTX (***p < 0.001, Figure 6C). In addition, the weight and overall survival period of mice administered with SynB3PVGLIGPTX (15 mg/5 mL kg-1, i.v.) were significantly higher than those in the control group (*p < 0.05, **p < 0.01, ***p < 0.001, Figure 6D,,E).E). Again, the separation in survival curves was notable among the four groups, with survival medians equal to 23, 29, and 25 for the control group, TMZ group and PTX group, respectively. In contrast, there was no death in the SynB3PVGLIGPTX group across the assays. Especially concerning was that, the administration concentration of SynB3PVGLIGPTX was 15 mg/5 mL kg-1, which is equivalent to 5.25 μmol/5 mL kg-1. Theoretically, after SynB3PVGLIGPTX at this concentration is completely hydrolyzed by MMP2, the concentration of free PTX released is 4.49 mg/5 mL kg-1, which is only onethird of the concentration administered in the PTX group. The experimental results show that, compared with the group given a high dose of PTX (15 mg/5 mL kg-1), the tumor volume of mice given the nanoconjugate (SynB3PVGLIGPTX) was significantly reduced, and its overall survival was also significantly prolonged, which indicates that SynB3PVGLIGPTX has a significant antiglioma activity in vivo. Accordingly, SynB3PVGLIGPTX, as a PDC that combined PTX to a dualfunctional peptide consisting of SynB3 and a MMP2sensitive peptide, could observably improve survival and decrease weight loss over the PTX monotherapy in vivo, and significantly overmatched the mice treated with TMZ.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c athymic nude mice xenograft model. | ||||
| In Vitro Model | Glioblastoma | U-87MG cell | CVCL_0022 | ||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [450] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 27.366 μM | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
SynB3PVGLIGPTX, was designed, screened, and synthetized, in which PTX was combined with CPPs (e.g., SynB3) through an MMP2sensitive linker (PVGLIG). This peptidedrug complex exhibits three advantages as follows: 1) the constructed structure of CPPs should help enhance the permeability of PTXcontaining nanocomplex across the BBB; 2) this nanocomplex covers a MMP2sensitive linker between CPPs and PTX, making it possible to release the drug at the target site with high MMP2 expression level; and 3) the novel dualfunctional PTX prodrug is a watersoluble nanocomplex, which can overcome the side effect exerted by low solubility and formulating agent (Cremophore EL) of PTX.
Click to Show/Hide
|
||||
| Description |
However, in the MMP2 siRNAtransfected U87MG and U251, SynB3PVGLIGPTX could result in an extremely lower inhibitory effect on cell proliferation than that treated at the same concentration ranges (25 x 10-6, 50 x 10-6, 75 x 10-6, and 100 x 10-6 M) in the U87MG and U251 cell lines (Figure 5G,,H).H). The absence of MMP2 manifested as an obviously reducing trend of IC50 of SynB3PVGLIGPTX in both cell lines (Table 3). These results indicated that SynB3PVGLIGPTX could only inhibit cell proliferation based on the presence of MMP2, which suggested that SynB3PVGLIGPTX performs a specific inhibitory action on the proliferation of GBM cells. In addition, in order to further verify the mechanism underlying the specific inhibitory activity of SynB3PVGLIGPTX on GBM cell proliferation, a supplementary experiment was performed using SynB3PTXtreated cells as inactive control groups. The results showed that the inhibition rates of SynB3PTX without an MMP2sensitive linker (PVGLIG) in U87MG and U251 were extremely low, at a nearzero level. Based on these observations, we confirmed that SynB3PVGLIGPTX has a specific antigrowth activity, that is, only in the presence of MMP2, the sensitive linker (PVGLIG) contained in SynB3PVGLIGPTX can be specifically hydrolyzed to release PTX, thereby inhibiting the proliferation of GBM cells.
Click to Show/Hide
|
||||
| In Vitro Model | Astrocytoma | U-251MG cell | CVCL_0021 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [450] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 31.981 μM | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
SynB3PVGLIGPTX, was designed, screened, and synthetized, in which PTX was combined with CPPs (e.g., SynB3) through an MMP2sensitive linker (PVGLIG). This peptidedrug complex exhibits three advantages as follows: 1) the constructed structure of CPPs should help enhance the permeability of PTXcontaining nanocomplex across the BBB; 2) this nanocomplex covers a MMP2sensitive linker between CPPs and PTX, making it possible to release the drug at the target site with high MMP2 expression level; and 3) the novel dualfunctional PTX prodrug is a watersoluble nanocomplex, which can overcome the side effect exerted by low solubility and formulating agent (Cremophore EL) of PTX.
Click to Show/Hide
|
||||
| Description |
However, in the MMP2 siRNAtransfected U87MG and U251, SynB3PVGLIGPTX could result in an extremely lower inhibitory effect on cell proliferation than that treated at the same concentration ranges (25 x 10-6, 50 x 10-6, 75 x 10-6, and 100 x 10-6 M) in the U87MG and U251 cell lines (Figure 5G,,H).H). The absence of MMP2 manifested as an obviously reducing trend of IC50 of SynB3PVGLIGPTX in both cell lines (Table 3). These results indicated that SynB3PVGLIGPTX could only inhibit cell proliferation based on the presence of MMP2, which suggested that SynB3PVGLIGPTX performs a specific inhibitory action on the proliferation of GBM cells. In addition, in order to further verify the mechanism underlying the specific inhibitory activity of SynB3PVGLIGPTX on GBM cell proliferation, a supplementary experiment was performed using SynB3PTXtreated cells as inactive control groups. The results showed that the inhibition rates of SynB3PTX without an MMP2sensitive linker (PVGLIG) in U87MG and U251 were extremely low, at a nearzero level. Based on these observations, we confirmed that SynB3PVGLIGPTX has a specific antigrowth activity, that is, only in the presence of MMP2, the sensitive linker (PVGLIG) contained in SynB3PVGLIGPTX can be specifically hydrolyzed to release PTX, thereby inhibiting the proliferation of GBM cells.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U-87MG cell | CVCL_0022 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [450] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 41.413 μM | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 24 h | ||||
| MOA of PDC |
SynB3PVGLIGPTX, was designed, screened, and synthetized, in which PTX was combined with CPPs (e.g., SynB3) through an MMP2sensitive linker (PVGLIG). This peptidedrug complex exhibits three advantages as follows: 1) the constructed structure of CPPs should help enhance the permeability of PTXcontaining nanocomplex across the BBB; 2) this nanocomplex covers a MMP2sensitive linker between CPPs and PTX, making it possible to release the drug at the target site with high MMP2 expression level; and 3) the novel dualfunctional PTX prodrug is a watersoluble nanocomplex, which can overcome the side effect exerted by low solubility and formulating agent (Cremophore EL) of PTX.
Click to Show/Hide
|
||||
| Description |
However, in the MMP2 siRNAtransfected U87MG and U251, SynB3PVGLIGPTX could result in an extremely lower inhibitory effect on cell proliferation than that treated at the same concentration ranges (25 x 10-6, 50 x 10-6, 75 x 10-6, and 100 x 10-6 M) in the U87MG and U251 cell lines (Figure 5G,,H).H). The absence of MMP2 manifested as an obviously reducing trend of IC50 of SynB3PVGLIGPTX in both cell lines (Table 3). These results indicated that SynB3PVGLIGPTX could only inhibit cell proliferation based on the presence of MMP2, which suggested that SynB3PVGLIGPTX performs a specific inhibitory action on the proliferation of GBM cells. In addition, in order to further verify the mechanism underlying the specific inhibitory activity of SynB3PVGLIGPTX on GBM cell proliferation, a supplementary experiment was performed using SynB3PTXtreated cells as inactive control groups. The results showed that the inhibition rates of SynB3PTX without an MMP2sensitive linker (PVGLIG) in U87MG and U251 were extremely low, at a nearzero level. Based on these observations, we confirmed that SynB3PVGLIGPTX has a specific antigrowth activity, that is, only in the presence of MMP2, the sensitive linker (PVGLIG) contained in SynB3PVGLIGPTX can be specifically hydrolyzed to release PTX, thereby inhibiting the proliferation of GBM cells.
Click to Show/Hide
|
||||
| In Vitro Model | Astrocytoma | U-251MG cell | CVCL_0021 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [450] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 45.787 μM | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 24 h | ||||
| MOA of PDC |
SynB3PVGLIGPTX, was designed, screened, and synthetized, in which PTX was combined with CPPs (e.g., SynB3) through an MMP2sensitive linker (PVGLIG). This peptidedrug complex exhibits three advantages as follows: 1) the constructed structure of CPPs should help enhance the permeability of PTXcontaining nanocomplex across the BBB; 2) this nanocomplex covers a MMP2sensitive linker between CPPs and PTX, making it possible to release the drug at the target site with high MMP2 expression level; and 3) the novel dualfunctional PTX prodrug is a watersoluble nanocomplex, which can overcome the side effect exerted by low solubility and formulating agent (Cremophore EL) of PTX.
Click to Show/Hide
|
||||
| Description |
However, in the MMP2 siRNAtransfected U87MG and U251, SynB3PVGLIGPTX could result in an extremely lower inhibitory effect on cell proliferation than that treated at the same concentration ranges (25 x 10-6, 50 x 10-6, 75 x 10-6, and 100 x 10-6 M) in the U87MG and U251 cell lines (Figure 5G,,H).H). The absence of MMP2 manifested as an obviously reducing trend of IC50 of SynB3PVGLIGPTX in both cell lines (Table 3). These results indicated that SynB3PVGLIGPTX could only inhibit cell proliferation based on the presence of MMP2, which suggested that SynB3PVGLIGPTX performs a specific inhibitory action on the proliferation of GBM cells. In addition, in order to further verify the mechanism underlying the specific inhibitory activity of SynB3PVGLIGPTX on GBM cell proliferation, a supplementary experiment was performed using SynB3PTXtreated cells as inactive control groups. The results showed that the inhibition rates of SynB3PTX without an MMP2sensitive linker (PVGLIG) in U87MG and U251 were extremely low, at a nearzero level. Based on these observations, we confirmed that SynB3PVGLIGPTX has a specific antigrowth activity, that is, only in the presence of MMP2, the sensitive linker (PVGLIG) contained in SynB3PVGLIGPTX can be specifically hydrolyzed to release PTX, thereby inhibiting the proliferation of GBM cells.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U-87MG cell | CVCL_0022 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [450] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 115.846 μM | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 4 h | ||||
| MOA of PDC |
SynB3PVGLIGPTX, was designed, screened, and synthetized, in which PTX was combined with CPPs (e.g., SynB3) through an MMP2sensitive linker (PVGLIG). This peptidedrug complex exhibits three advantages as follows: 1) the constructed structure of CPPs should help enhance the permeability of PTXcontaining nanocomplex across the BBB; 2) this nanocomplex covers a MMP2sensitive linker between CPPs and PTX, making it possible to release the drug at the target site with high MMP2 expression level; and 3) the novel dualfunctional PTX prodrug is a watersoluble nanocomplex, which can overcome the side effect exerted by low solubility and formulating agent (Cremophore EL) of PTX.
Click to Show/Hide
|
||||
| Description |
However, in the MMP2 siRNAtransfected U87MG and U251, SynB3PVGLIGPTX could result in an extremely lower inhibitory effect on cell proliferation than that treated at the same concentration ranges (25 x 10-6, 50 x 10-6, 75 x 10-6, and 100 x 10-6 M) in the U87MG and U251 cell lines (Figure 5G,,H).H). The absence of MMP2 manifested as an obviously reducing trend of IC50 of SynB3PVGLIGPTX in both cell lines (Table 3). These results indicated that SynB3PVGLIGPTX could only inhibit cell proliferation based on the presence of MMP2, which suggested that SynB3PVGLIGPTX performs a specific inhibitory action on the proliferation of GBM cells. In addition, in order to further verify the mechanism underlying the specific inhibitory activity of SynB3PVGLIGPTX on GBM cell proliferation, a supplementary experiment was performed using SynB3PTXtreated cells as inactive control groups. The results showed that the inhibition rates of SynB3PTX without an MMP2sensitive linker (PVGLIG) in U87MG and U251 were extremely low, at a nearzero level. Based on these observations, we confirmed that SynB3PVGLIGPTX has a specific antigrowth activity, that is, only in the presence of MMP2, the sensitive linker (PVGLIG) contained in SynB3PVGLIGPTX can be specifically hydrolyzed to release PTX, thereby inhibiting the proliferation of GBM cells.
Click to Show/Hide
|
||||
| In Vitro Model | Astrocytoma | U-251MG cell | CVCL_0021 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [450] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 139.541 μM | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 4 h | ||||
| MOA of PDC |
SynB3PVGLIGPTX, was designed, screened, and synthetized, in which PTX was combined with CPPs (e.g., SynB3) through an MMP2sensitive linker (PVGLIG). This peptidedrug complex exhibits three advantages as follows: 1) the constructed structure of CPPs should help enhance the permeability of PTXcontaining nanocomplex across the BBB; 2) this nanocomplex covers a MMP2sensitive linker between CPPs and PTX, making it possible to release the drug at the target site with high MMP2 expression level; and 3) the novel dualfunctional PTX prodrug is a watersoluble nanocomplex, which can overcome the side effect exerted by low solubility and formulating agent (Cremophore EL) of PTX.
Click to Show/Hide
|
||||
| Description |
However, in the MMP2 siRNAtransfected U87MG and U251, SynB3PVGLIGPTX could result in an extremely lower inhibitory effect on cell proliferation than that treated at the same concentration ranges (25 x 10-6, 50 x 10-6, 75 x 10-6, and 100 x 10-6 M) in the U87MG and U251 cell lines (Figure 5G,,H).H). The absence of MMP2 manifested as an obviously reducing trend of IC50 of SynB3PVGLIGPTX in both cell lines (Table 3). These results indicated that SynB3PVGLIGPTX could only inhibit cell proliferation based on the presence of MMP2, which suggested that SynB3PVGLIGPTX performs a specific inhibitory action on the proliferation of GBM cells. In addition, in order to further verify the mechanism underlying the specific inhibitory activity of SynB3PVGLIGPTX on GBM cell proliferation, a supplementary experiment was performed using SynB3PTXtreated cells as inactive control groups. The results showed that the inhibition rates of SynB3PTX without an MMP2sensitive linker (PVGLIG) in U87MG and U251 were extremely low, at a nearzero level. Based on these observations, we confirmed that SynB3PVGLIGPTX has a specific antigrowth activity, that is, only in the presence of MMP2, the sensitive linker (PVGLIG) contained in SynB3PVGLIGPTX can be specifically hydrolyzed to release PTX, thereby inhibiting the proliferation of GBM cells.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U-87MG cell | CVCL_0022 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [450] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 165.602 μM | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 4 h | ||||
| MOA of PDC |
SynB3PVGLIGPTX, was designed, screened, and synthetized, in which PTX was combined with CPPs (e.g., SynB3) through an MMP2sensitive linker (PVGLIG). This peptidedrug complex exhibits three advantages as follows: 1) the constructed structure of CPPs should help enhance the permeability of PTXcontaining nanocomplex across the BBB; 2) this nanocomplex covers a MMP2sensitive linker between CPPs and PTX, making it possible to release the drug at the target site with high MMP2 expression level; and 3) the novel dualfunctional PTX prodrug is a watersoluble nanocomplex, which can overcome the side effect exerted by low solubility and formulating agent (Cremophore EL) of PTX.
Click to Show/Hide
|
||||
| Description |
However, in the MMP2 siRNAtransfected U87MG and U251, SynB3PVGLIGPTX could result in an extremely lower inhibitory effect on cell proliferation than that treated at the same concentration ranges (25 x 10-6, 50 x 10-6, 75 x 10-6, and 100 x 10-6 M) in the U87MG and U251 cell lines (Figure 5G,,H).H). The absence of MMP2 manifested as an obviously reducing trend of IC50 of SynB3PVGLIGPTX in both cell lines (Table 3). These results indicated that SynB3PVGLIGPTX could only inhibit cell proliferation based on the presence of MMP2, which suggested that SynB3PVGLIGPTX performs a specific inhibitory action on the proliferation of GBM cells. In addition, in order to further verify the mechanism underlying the specific inhibitory activity of SynB3PVGLIGPTX on GBM cell proliferation, a supplementary experiment was performed using SynB3PTXtreated cells as inactive control groups. The results showed that the inhibition rates of SynB3PTX without an MMP2sensitive linker (PVGLIG) in U87MG and U251 were extremely low, at a nearzero level. Based on these observations, we confirmed that SynB3PVGLIGPTX has a specific antigrowth activity, that is, only in the presence of MMP2, the sensitive linker (PVGLIG) contained in SynB3PVGLIGPTX can be specifically hydrolyzed to release PTX, thereby inhibiting the proliferation of GBM cells.
Click to Show/Hide
|
||||
| In Vitro Model | Astrocytoma | U-251MG cell | CVCL_0021 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [450] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 172.180 μM | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
SynB3PVGLIGPTX, was designed, screened, and synthetized, in which PTX was combined with CPPs (e.g., SynB3) through an MMP2sensitive linker (PVGLIG). This peptidedrug complex exhibits three advantages as follows: 1) the constructed structure of CPPs should help enhance the permeability of PTXcontaining nanocomplex across the BBB; 2) this nanocomplex covers a MMP2sensitive linker between CPPs and PTX, making it possible to release the drug at the target site with high MMP2 expression level; and 3) the novel dualfunctional PTX prodrug is a watersoluble nanocomplex, which can overcome the side effect exerted by low solubility and formulating agent (Cremophore EL) of PTX.
Click to Show/Hide
|
||||
| Description |
However, in the MMP2 siRNAtransfected U87MG and U251, SynB3PVGLIGPTX could result in an extremely lower inhibitory effect on cell proliferation than that treated at the same concentration ranges (25 x 10-6, 50 x 10-6, 75 x 10-6, and 100 x 10-6 M) in the U87MG and U251 cell lines (Figure 5G,,H).H). The absence of MMP2 manifested as an obviously reducing trend of IC50 of SynB3PVGLIGPTX in both cell lines (Table 3). These results indicated that SynB3PVGLIGPTX could only inhibit cell proliferation based on the presence of MMP2, which suggested that SynB3PVGLIGPTX performs a specific inhibitory action on the proliferation of GBM cells. In addition, in order to further verify the mechanism underlying the specific inhibitory activity of SynB3PVGLIGPTX on GBM cell proliferation, a supplementary experiment was performed using SynB3PTXtreated cells as inactive control groups. The results showed that the inhibition rates of SynB3PTX without an MMP2sensitive linker (PVGLIG) in U87MG and U251 were extremely low, at a nearzero level. Based on these observations, we confirmed that SynB3PVGLIGPTX has a specific antigrowth activity, that is, only in the presence of MMP2, the sensitive linker (PVGLIG) contained in SynB3PVGLIGPTX can be specifically hydrolyzed to release PTX, thereby inhibiting the proliferation of GBM cells.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U-87MG cell | CVCL_0022 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [450] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 176.806 μM | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
SynB3PVGLIGPTX, was designed, screened, and synthetized, in which PTX was combined with CPPs (e.g., SynB3) through an MMP2sensitive linker (PVGLIG). This peptidedrug complex exhibits three advantages as follows: 1) the constructed structure of CPPs should help enhance the permeability of PTXcontaining nanocomplex across the BBB; 2) this nanocomplex covers a MMP2sensitive linker between CPPs and PTX, making it possible to release the drug at the target site with high MMP2 expression level; and 3) the novel dualfunctional PTX prodrug is a watersoluble nanocomplex, which can overcome the side effect exerted by low solubility and formulating agent (Cremophore EL) of PTX.
Click to Show/Hide
|
||||
| Description |
However, in the MMP2 siRNAtransfected U87MG and U251, SynB3PVGLIGPTX could result in an extremely lower inhibitory effect on cell proliferation than that treated at the same concentration ranges (25 x 10-6, 50 x 10-6, 75 x 10-6, and 100 x 10-6 M) in the U87MG and U251 cell lines (Figure 5G,,H).H). The absence of MMP2 manifested as an obviously reducing trend of IC50 of SynB3PVGLIGPTX in both cell lines (Table 3). These results indicated that SynB3PVGLIGPTX could only inhibit cell proliferation based on the presence of MMP2, which suggested that SynB3PVGLIGPTX performs a specific inhibitory action on the proliferation of GBM cells. In addition, in order to further verify the mechanism underlying the specific inhibitory activity of SynB3PVGLIGPTX on GBM cell proliferation, a supplementary experiment was performed using SynB3PTXtreated cells as inactive control groups. The results showed that the inhibition rates of SynB3PTX without an MMP2sensitive linker (PVGLIG) in U87MG and U251 were extremely low, at a nearzero level. Based on these observations, we confirmed that SynB3PVGLIGPTX has a specific antigrowth activity, that is, only in the presence of MMP2, the sensitive linker (PVGLIG) contained in SynB3PVGLIGPTX can be specifically hydrolyzed to release PTX, thereby inhibiting the proliferation of GBM cells.
Click to Show/Hide
|
||||
| In Vitro Model | Astrocytoma | U-251MG cell | CVCL_0021 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [450] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 207.674 μM | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 24 h | ||||
| MOA of PDC |
SynB3PVGLIGPTX, was designed, screened, and synthetized, in which PTX was combined with CPPs (e.g., SynB3) through an MMP2sensitive linker (PVGLIG). This peptidedrug complex exhibits three advantages as follows: 1) the constructed structure of CPPs should help enhance the permeability of PTXcontaining nanocomplex across the BBB; 2) this nanocomplex covers a MMP2sensitive linker between CPPs and PTX, making it possible to release the drug at the target site with high MMP2 expression level; and 3) the novel dualfunctional PTX prodrug is a watersoluble nanocomplex, which can overcome the side effect exerted by low solubility and formulating agent (Cremophore EL) of PTX.
Click to Show/Hide
|
||||
| Description |
However, in the MMP2 siRNAtransfected U87MG and U251, SynB3PVGLIGPTX could result in an extremely lower inhibitory effect on cell proliferation than that treated at the same concentration ranges (25 x 10-6, 50 x 10-6, 75 x 10-6, and 100 x 10-6 M) in the U87MG and U251 cell lines (Figure 5G,,H).H). The absence of MMP2 manifested as an obviously reducing trend of IC50 of SynB3PVGLIGPTX in both cell lines (Table 3). These results indicated that SynB3PVGLIGPTX could only inhibit cell proliferation based on the presence of MMP2, which suggested that SynB3PVGLIGPTX performs a specific inhibitory action on the proliferation of GBM cells. In addition, in order to further verify the mechanism underlying the specific inhibitory activity of SynB3PVGLIGPTX on GBM cell proliferation, a supplementary experiment was performed using SynB3PTXtreated cells as inactive control groups. The results showed that the inhibition rates of SynB3PTX without an MMP2sensitive linker (PVGLIG) in U87MG and U251 were extremely low, at a nearzero level. Based on these observations, we confirmed that SynB3PVGLIGPTX has a specific antigrowth activity, that is, only in the presence of MMP2, the sensitive linker (PVGLIG) contained in SynB3PVGLIGPTX can be specifically hydrolyzed to release PTX, thereby inhibiting the proliferation of GBM cells.
Click to Show/Hide
|
||||
| In Vitro Model | Astrocytoma | U-251MG cell | CVCL_0021 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [450] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 219.364 μM | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 24 h | ||||
| MOA of PDC |
SynB3PVGLIGPTX, was designed, screened, and synthetized, in which PTX was combined with CPPs (e.g., SynB3) through an MMP2sensitive linker (PVGLIG). This peptidedrug complex exhibits three advantages as follows: 1) the constructed structure of CPPs should help enhance the permeability of PTXcontaining nanocomplex across the BBB; 2) this nanocomplex covers a MMP2sensitive linker between CPPs and PTX, making it possible to release the drug at the target site with high MMP2 expression level; and 3) the novel dualfunctional PTX prodrug is a watersoluble nanocomplex, which can overcome the side effect exerted by low solubility and formulating agent (Cremophore EL) of PTX.
Click to Show/Hide
|
||||
| Description |
However, in the MMP2 siRNAtransfected U87MG and U251, SynB3PVGLIGPTX could result in an extremely lower inhibitory effect on cell proliferation than that treated at the same concentration ranges (25 x 10-6, 50 x 10-6, 75 x 10-6, and 100 x 10-6 M) in the U87MG and U251 cell lines (Figure 5G,,H).H). The absence of MMP2 manifested as an obviously reducing trend of IC50 of SynB3PVGLIGPTX in both cell lines (Table 3). These results indicated that SynB3PVGLIGPTX could only inhibit cell proliferation based on the presence of MMP2, which suggested that SynB3PVGLIGPTX performs a specific inhibitory action on the proliferation of GBM cells. In addition, in order to further verify the mechanism underlying the specific inhibitory activity of SynB3PVGLIGPTX on GBM cell proliferation, a supplementary experiment was performed using SynB3PTXtreated cells as inactive control groups. The results showed that the inhibition rates of SynB3PTX without an MMP2sensitive linker (PVGLIG) in U87MG and U251 were extremely low, at a nearzero level. Based on these observations, we confirmed that SynB3PVGLIGPTX has a specific antigrowth activity, that is, only in the presence of MMP2, the sensitive linker (PVGLIG) contained in SynB3PVGLIGPTX can be specifically hydrolyzed to release PTX, thereby inhibiting the proliferation of GBM cells.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U-87MG cell | CVCL_0022 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [450] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 311.219 μM | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 4 h | ||||
| MOA of PDC |
SynB3PVGLIGPTX, was designed, screened, and synthetized, in which PTX was combined with CPPs (e.g., SynB3) through an MMP2sensitive linker (PVGLIG). This peptidedrug complex exhibits three advantages as follows: 1) the constructed structure of CPPs should help enhance the permeability of PTXcontaining nanocomplex across the BBB; 2) this nanocomplex covers a MMP2sensitive linker between CPPs and PTX, making it possible to release the drug at the target site with high MMP2 expression level; and 3) the novel dualfunctional PTX prodrug is a watersoluble nanocomplex, which can overcome the side effect exerted by low solubility and formulating agent (Cremophore EL) of PTX.
Click to Show/Hide
|
||||
| Description |
However, in the MMP2 siRNAtransfected U87MG and U251, SynB3PVGLIGPTX could result in an extremely lower inhibitory effect on cell proliferation than that treated at the same concentration ranges (25 x 10-6, 50 x 10-6, 75 x 10-6, and 100 x 10-6 M) in the U87MG and U251 cell lines (Figure 5G,,H).H). The absence of MMP2 manifested as an obviously reducing trend of IC50 of SynB3PVGLIGPTX in both cell lines (Table 3). These results indicated that SynB3PVGLIGPTX could only inhibit cell proliferation based on the presence of MMP2, which suggested that SynB3PVGLIGPTX performs a specific inhibitory action on the proliferation of GBM cells. In addition, in order to further verify the mechanism underlying the specific inhibitory activity of SynB3PVGLIGPTX on GBM cell proliferation, a supplementary experiment was performed using SynB3PTXtreated cells as inactive control groups. The results showed that the inhibition rates of SynB3PTX without an MMP2sensitive linker (PVGLIG) in U87MG and U251 were extremely low, at a nearzero level. Based on these observations, we confirmed that SynB3PVGLIGPTX has a specific antigrowth activity, that is, only in the presence of MMP2, the sensitive linker (PVGLIG) contained in SynB3PVGLIGPTX can be specifically hydrolyzed to release PTX, thereby inhibiting the proliferation of GBM cells.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U-87MG cell | CVCL_0022 | ||
ANG-TAT-PTX [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [451] | ||||
| Indication | Central nervous system disease | ||||
| Efficacy Data | Inhinition rate | 31.65 ± 3.28% | |||
| Administration Time | 24 h | ||||
| In Vitro Model | Glioblastoma | U87 cell | CVCL_3429 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [451] | ||||
| Indication | Central nervous system disease | ||||
| Efficacy Data | Inhinition rate | 73.53 ± 6.45% | |||
| Administration Time | 48 h | ||||
| In Vitro Model | Glioblastoma | U87 cell | CVCL_3429 | ||
ANG-PTX [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [451] | ||||
| Indication | Central nervous system disease | ||||
| Efficacy Data | Inhinition rate | 33.21 ± 3.32% | |||
| Administration Time | 24 h | ||||
| In Vitro Model | Glioblastoma | U87 cell | CVCL_3429 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [451] | ||||
| Indication | Central nervous system disease | ||||
| Efficacy Data | Inhinition rate | 50.24 ± 4.75% | |||
| Administration Time | 48 h | ||||
| In Vitro Model | Glioblastoma | U87 cell | CVCL_3429 | ||
mPEG-AAN-PTP-7-CDM-PTX [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.044 µg/mL | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 72 h | ||||
| MOA of PDC |
Herein, we present a lytic peptide PTP7-drug paclitaxel conjugate assembling nanoparticles (named PPP) that can sequentially respond to dual stimuli in the tumor microenvironment, which was designed for passive tumor-targeted delivery and on-demand release of a tumor lytic peptide (PTP-7) as well as a chemotherapeutic agent of paclitaxel (PTX). To achieve this, tumor lytic peptide PTP-7 was connected with polyethylene glycol by a peptide substrate of legumain to serve as hydrophobic segments of nanoparticles to protect the peptide from enzymatic degradation. After that, PTX was connected to the amino group of the polypeptide side chain through an acid-responsive chemical bond (2-propionic-3-methylmaleic anhydride, CDM). Therefore, the nanoparticle (PPP) collapsed when it encountered the weakly acidic tumor microenvironment where PTX molecules fell off, and further triggered the cleavage of the peptide substrate by legumain that is highly expressed in tumor stroma and tumor cell surface. Moreover, PPP presents improved stability, improved drug solubility, prolonged blood circulation and significant inhibition ability on tumor growth, which gives a reasonable strategy to accurately deliver small molecule drugs and active peptides simultaneously to tumor sites.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of PPP in MCF-7, HCT116, and 4T1 cells was investigated by MTT assay. The half maximal inhibitory concentration (IC50) of PPP NPs and free PTX was calculated to be 0.066 and 0.028 μg/mL on MCF-7 cells, 0.044 and 0.006 μg/mL on HCT116, 0.212 and 0.01 μg/mL on 4T1 cells which indicated a lower cell growth inhibition ability than free PTX. The result may be owing to the extremely stable property of PPP and low legumain expression in vitro leading to incomplete release of PTX and PTP-7 (Edgington et al., 2013). Moreover, the holographic microscopy studies showed the changes in cellular morphology of MCF-7 cells after treated with PPP for 3 h. Notably, PPP led to the morphological features of apoptosis such as shrinkage, losing contact with neighboring cells and floating relative to control.
Click to Show/Hide
|
||||
| In Vitro Model | Colon carcinoma | HCT 116 cell | CVCL_0291 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.066 µg/mL | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 72 h | ||||
| MOA of PDC |
Herein, we present a lytic peptide PTP7-drug paclitaxel conjugate assembling nanoparticles (named PPP) that can sequentially respond to dual stimuli in the tumor microenvironment, which was designed for passive tumor-targeted delivery and on-demand release of a tumor lytic peptide (PTP-7) as well as a chemotherapeutic agent of paclitaxel (PTX). To achieve this, tumor lytic peptide PTP-7 was connected with polyethylene glycol by a peptide substrate of legumain to serve as hydrophobic segments of nanoparticles to protect the peptide from enzymatic degradation. After that, PTX was connected to the amino group of the polypeptide side chain through an acid-responsive chemical bond (2-propionic-3-methylmaleic anhydride, CDM). Therefore, the nanoparticle (PPP) collapsed when it encountered the weakly acidic tumor microenvironment where PTX molecules fell off, and further triggered the cleavage of the peptide substrate by legumain that is highly expressed in tumor stroma and tumor cell surface. Moreover, PPP presents improved stability, improved drug solubility, prolonged blood circulation and significant inhibition ability on tumor growth, which gives a reasonable strategy to accurately deliver small molecule drugs and active peptides simultaneously to tumor sites.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of PPP in MCF-7, HCT116, and 4T1 cells was investigated by MTT assay. The half maximal inhibitory concentration (IC50) of PPP NPs and free PTX was calculated to be 0.066 and 0.028 μg/mL on MCF-7 cells, 0.044 and 0.006 μg/mL on HCT116, 0.212 and 0.01 μg/mL on 4T1 cells which indicated a lower cell growth inhibition ability than free PTX. The result may be owing to the extremely stable property of PPP and low legumain expression in vitro leading to incomplete release of PTX and PTP-7 (Edgington et al., 2013). Moreover, the holographic microscopy studies showed the changes in cellular morphology of MCF-7 cells after treated with PPP for 3 h. Notably, PPP led to the morphological features of apoptosis such as shrinkage, losing contact with neighboring cells and floating relative to control.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.212 µg/mL | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 72 h | ||||
| MOA of PDC |
Herein, we present a lytic peptide PTP7-drug paclitaxel conjugate assembling nanoparticles (named PPP) that can sequentially respond to dual stimuli in the tumor microenvironment, which was designed for passive tumor-targeted delivery and on-demand release of a tumor lytic peptide (PTP-7) as well as a chemotherapeutic agent of paclitaxel (PTX). To achieve this, tumor lytic peptide PTP-7 was connected with polyethylene glycol by a peptide substrate of legumain to serve as hydrophobic segments of nanoparticles to protect the peptide from enzymatic degradation. After that, PTX was connected to the amino group of the polypeptide side chain through an acid-responsive chemical bond (2-propionic-3-methylmaleic anhydride, CDM). Therefore, the nanoparticle (PPP) collapsed when it encountered the weakly acidic tumor microenvironment where PTX molecules fell off, and further triggered the cleavage of the peptide substrate by legumain that is highly expressed in tumor stroma and tumor cell surface. Moreover, PPP presents improved stability, improved drug solubility, prolonged blood circulation and significant inhibition ability on tumor growth, which gives a reasonable strategy to accurately deliver small molecule drugs and active peptides simultaneously to tumor sites.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of PPP in MCF-7, HCT116, and 4T1 cells was investigated by MTT assay. The half maximal inhibitory concentration (IC50) of PPP NPs and free PTX was calculated to be 0.066 and 0.028 μg/mL on MCF-7 cells, 0.044 and 0.006 μg/mL on HCT116, 0.212 and 0.01 μg/mL on 4T1 cells which indicated a lower cell growth inhibition ability than free PTX. The result may be owing to the extremely stable property of PPP and low legumain expression in vitro leading to incomplete release of PTX and PTP-7 (Edgington et al., 2013). Moreover, the holographic microscopy studies showed the changes in cellular morphology of MCF-7 cells after treated with PPP for 3 h. Notably, PPP led to the morphological features of apoptosis such as shrinkage, losing contact with neighboring cells and floating relative to control.
Click to Show/Hide
|
||||
| In Vitro Model | Mammary carcinoma | 4T1 cell | CVCL_0125 | ||
GnRH-III-[2ΔHis-3D-Tic, 8Lys(glutaryl-Val-Ala-PABC-diamine-PTX)] conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.51 ± 0.11 µM | |||
| Evaluation Method | GraphPad prism assay | ||||
| Administration Time | 72 h | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.67 ± 0.07 µM | |||
| Evaluation Method | GraphPad prism assay | ||||
| Administration Time | 72 h | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 5.03 ± 1.91 µM | |||
| Evaluation Method | GraphPad prism assay | ||||
| Administration Time | 72 h | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | PANC-1 cell | CVCL_0480 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 6.44 ± 1.22 µM | |||
| Evaluation Method | GraphPad prism assay | ||||
| Administration Time | 72 h | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | PANC-1 cell | CVCL_0480 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 10.54 ± 2.01 µM | |||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Normal human pituitary cell | Homo sapiens | ||
| Experiment 6 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 10.65 ± 1.82 µM | |||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate cancer | Human prostate cancer cells | Homo sapiens | ||
| Experiment 7 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 14.88 ± 0.33 µM | |||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate cancer | Human prostate cancer cells | Homo sapiens | ||
| Experiment 8 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 18.49 ± 2.72 µM | |||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Normal human pituitary cell | Homo sapiens | ||
GnRH-III-[2His-3Trp,8Lys(glutaryl-Dau)] conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.66 ± 0.18 µM | |||
| Evaluation Method | GraphPad prism assay | ||||
| Administration Time | 72 h | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 4.89 ± 1.08 µM | |||
| Evaluation Method | GraphPad prism assay | ||||
| Administration Time | 72 h | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | PANC-1 cell | CVCL_0480 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 8.47 ± 1.06 µM | |||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate cancer | Human prostate cancer cells | Homo sapiens | ||
| Experiment 4 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 9.86 ± 0.82 µM | |||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Normal human pituitary cell | Homo sapiens | ||
GnRH-III-[2ΔHis-3D-Tic,8Lys(glutaryl-Dau)] conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.77 ± 0.08 µM | |||
| Evaluation Method | GraphPad prism assay | ||||
| Administration Time | 72 h | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 8.15 ± 3.22 µM | |||
| Evaluation Method | GraphPad prism assay | ||||
| Administration Time | 72 h | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | PANC-1 cell | CVCL_0480 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 10.82 ± 1.98 µM | |||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Normal human pituitary cell | Homo sapiens | ||
| Experiment 4 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 12.73 ± 2.23 µM | |||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate cancer | Human prostate cancer cells | Homo sapiens | ||
FA-P3-PTX [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [446] | ||||
| Indication | Invasive breast carcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 1.79 ± 0.09 μM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [446] | ||||
| Indication | Ovarian endometrioid adenocarcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 1.95 ± 0.20 μM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [446] | ||||
| Indication | Invasive ductal carcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 4.54 ± 0.71 μM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive ductal carcinoma | MCF7/PTX cell | CVCL_C5RS | ||
| Experiment 4 Reporting the Activity Data of This PDC | [446] | ||||
| Indication | Chronic myeloid leukemia | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 5.38 ± 0.25 μM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Chronic myeloid leukemia | K562 cell | CVCL_0004 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [446] | ||||
| Indication | Ovarian serous cystadenocarcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 5.92 ± 0.84 μM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [446] | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 48.55 ± 2.94 μM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Human umbilical vein endothelial cells | Homo sapiens | ||
Lytic peptides 6 - Paclitaxel conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [446] | ||||
| Indication | Invasive breast carcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 1.98 ± 0.14 μM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [446] | ||||
| Indication | Ovarian endometrioid adenocarcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 2.79 ± 0.17 μM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [446] | ||||
| Indication | Chronic myeloid leukemia | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 3.82 ± 0.29 μM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Chronic myeloid leukemia | K562 cell | CVCL_0004 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [446] | ||||
| Indication | Invasive ductal carcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 5.53 ± 0.76 μM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive ductal carcinoma | MCF7/PTX cell | CVCL_C5RS | ||
| Experiment 5 Reporting the Activity Data of This PDC | [446] | ||||
| Indication | Ovarian serous cystadenocarcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 6.61 ± 0.94 μM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [446] | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 37.22 ± 2.36 μM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Human umbilical vein endothelial cells | Homo sapiens | ||
Lytic peptides 5 - Paclitaxel conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [446] | ||||
| Indication | Invasive breast carcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 2.15 ± 0.18 μM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [446] | ||||
| Indication | Ovarian endometrioid adenocarcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 2.69 ± 0.19 μM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [446] | ||||
| Indication | Chronic myeloid leukemia | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 5.90 ± 0.92 μM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Chronic myeloid leukemia | K562 cell | CVCL_0004 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [446] | ||||
| Indication | Invasive ductal carcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 6.11 ± 0.61 μM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive ductal carcinoma | MCF7/PTX cell | CVCL_C5RS | ||
| Experiment 5 Reporting the Activity Data of This PDC | [446] | ||||
| Indication | Ovarian serous cystadenocarcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 7.17 ± 0.77 μM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [446] | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 31.60 ± 1.88 μM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Human umbilical vein endothelial cells | Homo sapiens | ||
GnRH-III-[2ΔHis-3D-Tic, 8Lys(glutaryl-diamine-PTX)] conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 22.21 ± 0.96 µM | |||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate cancer | Human prostate cancer cells | Homo sapiens | ||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 23.43 ± 1.67 µM | |||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Normal human pituitary cell | Homo sapiens | ||
| Experiment 3 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 100 µM | |||
| Evaluation Method | GraphPad prism assay | ||||
| Administration Time | 72 h | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 100 µM | |||
| Evaluation Method | GraphPad prism assay | ||||
| Administration Time | 72 h | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | PANC-1 cell | CVCL_0480 | ||
GnRH-III-[2His-3Trp,8Lys(glutaryl-diamine-PTX)] conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 41.52 ± 9.83 µM | |||
| Evaluation Method | GraphPad prism assay | ||||
| Administration Time | 72 h | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 100 µM | |||
| Evaluation Method | GraphPad prism assay | ||||
| Administration Time | 72 h | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | PANC-1 cell | CVCL_0480 | ||
CPP-SA-PTX [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Malignant glioblastoma | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) | 20.55 ± 1.02 nM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 24 h | ||||
| MOA of PDC |
A highly sensitive, nontoxic, hydrophilic cell-penetrating peptide (CPP = c[RGDKLAK]) was selected for the construction of an effective peptide-drug conjugate (PDC). A hydrophobic drug paclitaxel (PTX) was successfully conjugated with CPP via ester linkage with succinic acid (SA) as a pH-cleavable linker moiety. The characterization techniques employed in this study indicate the >95% purity of the resulting PDC (CPP-SA-PTX). The in vitro studies show that our proposed PDC exhibits enhanced stability (˜90%) and cytotoxicity (EC50 = 8.32 ± 0.09 nM). Besides the excellent solubility of PDC in water, the PTX effect on positive β-tubulin-III indicates that the drug releases retained pharmacological properties. Additionally, in vivo, therapeutic-dose treatment reveals the prominent tumor-growth inhibitory effects (2.82-3.24-fold) of PDC in tumor mice models. Subsequently, these observations confirmed that our novel-designed PDC (CPP-SA-PTX) adduct may serve as a promising therapeutic agent to treat glioblastoma.
Click to Show/Hide
|
||||
| Description |
Furthermore, it was also observed that PDC shows time-dependent cytotoxicity against U87MG cells, as at 12 h drug treatment. PDC shows a slightly lower inhibitory effect on cell survival (EC50 = 25.82 ± 0.31 nM) than PTX alone (EC50 = 12.25 ± 0.13 nM), and CPP-SA alone shows higher EC50 = 54.37 ± 0.24 in U87MG cells. Besides this, it was previously observed that CPP-treated healthy VERO cells showed 88.05 ± 0.86% viable cells even using 10 uM concentration, indicating the specificity of CPP for glioblastoma cells. However, at 24 h incubation period, our PDC shows a significantly enhanced cell survival inhibitory effect with EC50 = 8.32 ± 0.09 nM, suggesting the delayed effect of PDC because of the time required for the intracellular pH to cause maximal cleavage of PTX from the PDC. Moreover, it also shows that CPP-SA has a lower cytotoxicity effect on glioblastoma cells compared with PTX alone and PDC, indicating its specificity as a carrier and selectivity for integrin receptors overexpressed on the cell surface. Additionally, we have also studied the potential of our novel-designed PDC to internalize into PTX-resistant glioblastoma (U87MG-PR) cells to show cytotoxic activity upon intracellular cleavage of PTX from CPP. The data presented in Figure 3b reveal approximately 15% viability increases in U87MG-PR cells compared with parent U87MG cells. It can be seen in the inset in Figure 3b that PTX alone (EC50 = 41.3 ± 1.5 nM) showed significantly 2-fold reduced cytotoxic activity in U87MG-PR cells in comparison with our novel-designed PDC having EC50 = 20.55 ± 1.02 nM. It was also observed in Figure 3b that at lower concentrations (0-10 nM) the viable cell count was ˜75%; however, it significantly decreased to ˜20% viability with an increase in concentration (40 nM) of the test samples, highlighting the dose-dependent cytotoxicity behavior of PDC in a 24 h incubation period.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U87MG-PR cell | CVCL_0022 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Malignant glioblastoma | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) | 25.82 ± 0.31 nM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 24 h | ||||
| MOA of PDC |
A highly sensitive, nontoxic, hydrophilic cell-penetrating peptide (CPP = c[RGDKLAK]) was selected for the construction of an effective peptide-drug conjugate (PDC). A hydrophobic drug paclitaxel (PTX) was successfully conjugated with CPP via ester linkage with succinic acid (SA) as a pH-cleavable linker moiety. The characterization techniques employed in this study indicate the >95% purity of the resulting PDC (CPP-SA-PTX). The in vitro studies show that our proposed PDC exhibits enhanced stability (˜90%) and cytotoxicity (EC50 = 8.32 ± 0.09 nM). Besides the excellent solubility of PDC in water, the PTX effect on positive β-tubulin-III indicates that the drug releases retained pharmacological properties. Additionally, in vivo, therapeutic-dose treatment reveals the prominent tumor-growth inhibitory effects (2.82-3.24-fold) of PDC in tumor mice models. Subsequently, these observations confirmed that our novel-designed PDC (CPP-SA-PTX) adduct may serve as a promising therapeutic agent to treat glioblastoma.
Click to Show/Hide
|
||||
| Description |
Furthermore, it was also observed that PDC shows time-dependent cytotoxicity against U87MG cells, as at 12 h drug treatment. PDC shows a slightly lower inhibitory effect on cell survival (EC50 = 25.82 ± 0.31 nM) than PTX alone (EC50 = 12.25 ± 0.13 nM), and CPP-SA alone shows higher EC50 = 54.37 ± 0.24 in U87MG cells. Besides this, it was previously observed that CPP-treated healthy VERO cells showed 88.05 ± 0.86% viable cells even using 10 uM concentration, indicating the specificity of CPP for glioblastoma cells. However, at 24 h incubation period, our PDC shows a significantly enhanced cell survival inhibitory effect with EC50 = 8.32 ± 0.09 nM, suggesting the delayed effect of PDC because of the time required for the intracellular pH to cause maximal cleavage of PTX from the PDC. Moreover, it also shows that CPP-SA has a lower cytotoxicity effect on glioblastoma cells compared with PTX alone and PDC, indicating its specificity as a carrier and selectivity for integrin receptors overexpressed on the cell surface. Additionally, we have also studied the potential of our novel-designed PDC to internalize into PTX-resistant glioblastoma (U87MG-PR) cells to show cytotoxic activity upon intracellular cleavage of PTX from CPP. The data presented in Figure 3b reveal approximately 15% viability increases in U87MG-PR cells compared with parent U87MG cells. It can be seen in the inset in Figure 3b that PTX alone (EC50 = 41.3 ± 1.5 nM) showed significantly 2-fold reduced cytotoxic activity in U87MG-PR cells in comparison with our novel-designed PDC having EC50 = 20.55 ± 1.02 nM. It was also observed in Figure 3b that at lower concentrations (0-10 nM) the viable cell count was ˜75%; however, it significantly decreased to ˜20% viability with an increase in concentration (40 nM) of the test samples, highlighting the dose-dependent cytotoxicity behavior of PDC in a 24 h incubation period.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U-87MG cell | CVCL_0022 | ||
PDC-PTX1 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [452] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 60% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 5 µM | ||||
| Description |
Antiproliferative results showed that PTX1 inhibited cell proliferation by 18.7%. The anti-proliferative activity of CPT1 was diminished by 1.9-fold as compared to CPT whereas the activity of CPT2 was comparable to CPT, since CPT2 reduced the cell viability to 61%.
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [452] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 80% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 5 µM | ||||
| Description |
The cytotoxicity of PTX and PTX1 was further evaluated in the normal human embryonic kidney cells (HEK-293) at 5 uM which showed reduced cell proliferation by ~34% and 18%, respectively, after 72 h using MTT assay, as shown in Figure 2.
|
||||
| In Vitro Model | Normal | HEK-298 cell | Homo sapiens | ||
References