
Linker Information
General Information of This Linker
| Linker ID |
LIN00003
|
|||||
|---|---|---|---|---|---|---|
| Linker Name |
Succinic Acid
|
|||||
| Linker Type |
PH-Sensitive linkers
|
|||||
| Structure |
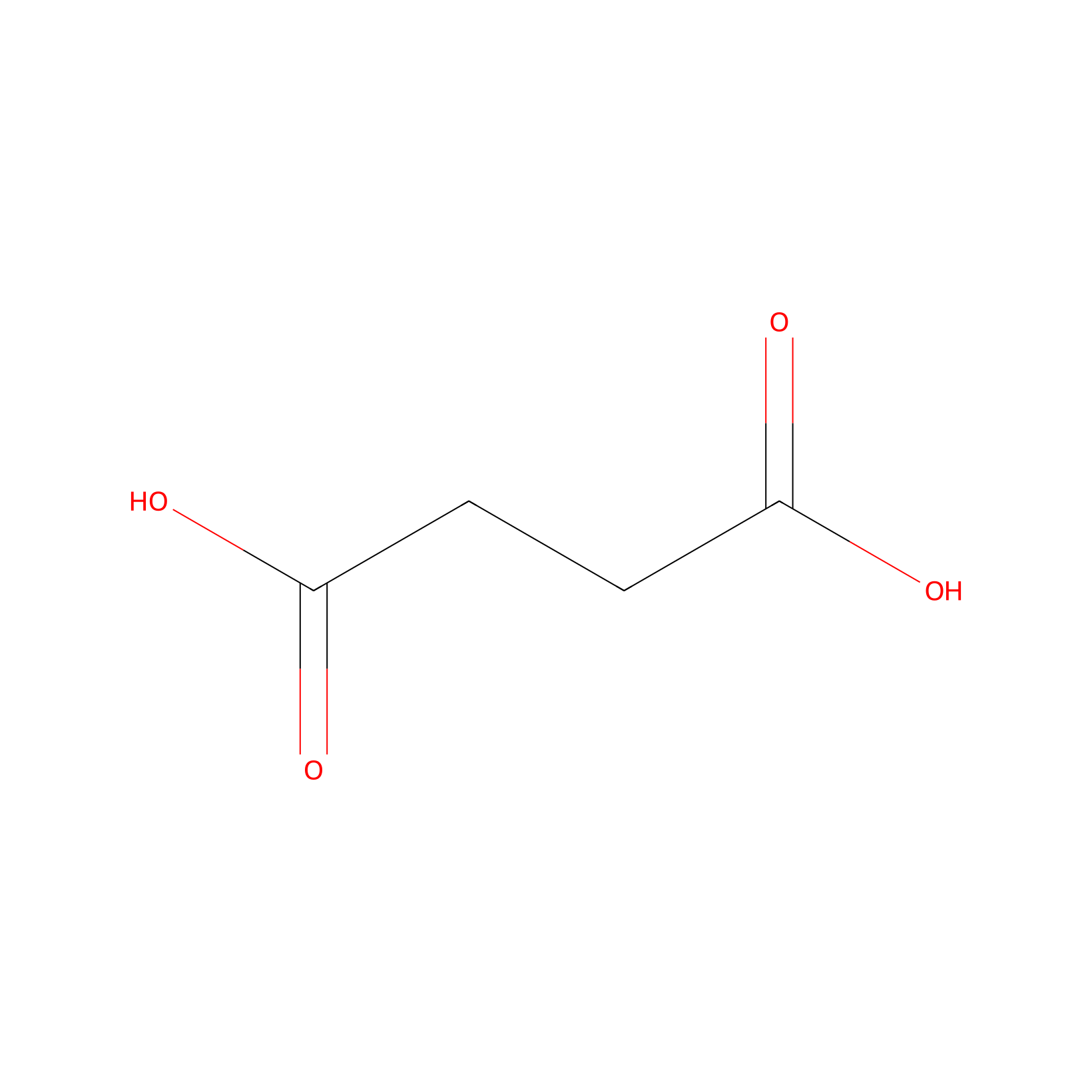
|
|||||
| Formula |
C4H6O4
|
|||||
| #Ro5 Violations (Lipinski): 0 | Molecular Weight (mw) | 118.09 | ||||
| Lipid-water partition coefficient (xlogp) | -0.6 | |||||
| Hydrogen Bond Donor Count (hbonddonor) | 2 | |||||
| Hydrogen Bond Acceptor Count (hbondacc) | 4 | |||||
| Rotatable Bond Count (rotbonds) | 3 | |||||
| Chemble ID | ||||||
| Chemble ID | ||||||
| PubChem CID | ||||||
| Canonical smiles |
C(CC(=O)O)C(=O)O
|
|||||
| InChI |
InChI=1S/C4H6O4/c5-3(6)1-2-4(7)8/h1-2H2,(H,5,6)(H,7,8)
|
|||||
| InChIKey |
KDYFGRWQOYBRFD-UHFFFAOYSA-N
|
|||||
| IUPAC Name |
butanedioic acid
|
|||||
Each Peptide-drug Conjugate Related to This Linker
Full Information of The Activity Data of The PDC(s) Related to This Linker
ANG1005 [Phase 3]
Identified from the Human Clinical Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Objective response rate (ORR) |
15%
|
|||
| Patients Enrolled |
Adult patients with measurable recurrent brain metastases from breast cancer.
|
||||
| Administration Time | Every 3 weeks | ||||
| Administration Dosage | 600 mg/m2 | ||||
| MOA of PDC |
Because LRP1 is also expressed on tumor cells in both CNS and systemic metastases, ANG1005 gains entry via LRP1 mediated endocytosis, where paclitaxel is cleaved from the peptide backbone by lysosomal esterases.
|
||||
| Description |
On the basis of the CNS tumor response assessment, performed by local investigators, there were nine (15%) evaluable patients with PR including five (8%) confirmed PR (to confirm PR, it was required that the response was sustained for ≥4 weeks), and 32 (53%) evaluable patients with SD, resulting in an overall iORR of 15% and iCBR of 68%.
|
||||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Objective response rate (ORR) |
29%
|
|||
| Patients Enrolled |
Patients with leptomeningeal carcinomatosis.
|
||||
| Administration Time | Every 3 weeks | ||||
| Administration Dosage | 600 mg/m2 | ||||
| MOA of PDC |
Because LRP1 is also expressed on tumor cells in both CNS and systemic metastases, ANG1005 gains entry via LRP1 mediated endocytosis, where paclitaxel is cleaved from the peptide backbone by lysosomal esterases.
|
||||
| Description |
Investigator determined ORR was 29% and the iCBR was 67%.
|
||||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
2.8 months
|
|||
| Patients Enrolled |
Patients with leptomeningeal carcinomatosis.
|
||||
| Administration Time | Every 3 weeks | ||||
| Administration Dosage | 600 mg/m2 | ||||
| MOA of PDC |
Because LRP1 is also expressed on tumor cells in both CNS and systemic metastases, ANG1005 gains entry via LRP1 mediated endocytosis, where paclitaxel is cleaved from the peptide backbone by lysosomal esterases.
|
||||
| Description |
The investigator determined intracranial median PFS was 2.8 months and the 3-month PFS rate was 54% (Table 3). Median duration of response was 18 weeks (7.3-26.3).
|
||||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
12.1 weeks
|
|||
| Patients Enrolled |
Adult patients with measurable recurrent brain metastases from breast cancer.
|
||||
| Administration Time | Every 3 weeks | ||||
| Administration Dosage | 600 mg/m2 | ||||
| MOA of PDC |
Because LRP1 is also expressed on tumor cells in both CNS and systemic metastases, ANG1005 gains entry via LRP1 mediated endocytosis, where paclitaxel is cleaved from the peptide backbone by lysosomal esterases.
|
||||
| Description |
Investigator assessments resulted in median intracranial PFS of 2.8 months and the 3-month intracranial PFS rate was 52%.
|
||||
| Experiment 5 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Median duration of response |
18 weeks
|
|||
| Patients Enrolled |
Patients with leptomeningeal carcinomatosis.
|
||||
| Administration Time | Every 3 weeks | ||||
| Administration Dosage | 600 mg/m2 | ||||
| MOA of PDC |
Because LRP1 is also expressed on tumor cells in both CNS and systemic metastases, ANG1005 gains entry via LRP1 mediated endocytosis, where paclitaxel is cleaved from the peptide backbone by lysosomal esterases.
|
||||
| Description |
The investigator determined intracranial median PFS was 2.8 months and the 3-month PFS rate was 54% (Table 3). Median duration of response was 18 weeks (7.3-26.3).
|
||||
| Experiment 6 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Clinical benefit rate (CBR) |
67%
|
|||
| Patients Enrolled |
Patients with leptomeningeal carcinomatosis.
|
||||
| Administration Time | Every 3 weeks | ||||
| Administration Dosage | 600 mg/m2 | ||||
| MOA of PDC |
Because LRP1 is also expressed on tumor cells in both CNS and systemic metastases, ANG1005 gains entry via LRP1 mediated endocytosis, where paclitaxel is cleaved from the peptide backbone by lysosomal esterases.
|
||||
| Description |
Investigator determined ORR was 29% and the iCBR was 67%.
|
||||
| Experiment 7 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Clinical benefit rate (CBR) |
68%
|
|||
| Patients Enrolled |
Adult patients with measurable recurrent brain metastases from breast cancer.
|
||||
| Administration Time | Every 3 weeks | ||||
| Administration Dosage | 600 mg/m2 | ||||
| MOA of PDC |
Because LRP1 is also expressed on tumor cells in both CNS and systemic metastases, ANG1005 gains entry via LRP1 mediated endocytosis, where paclitaxel is cleaved from the peptide backbone by lysosomal esterases.
|
||||
| Description |
On the basis of the CNS tumor response assessment, performed by local investigators, there were nine (15%) evaluable patients with PR including five (8%) confirmed PR (to confirm PR, it was required that the response was sustained for ≥4 weeks), and 32 (53%) evaluable patients with SD, resulting in an overall iORR of 15% and iCBR of 68%.
|
||||
| Experiment 8 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | 6-month progression-free survival rate |
18.70%
|
|||
| Patients Enrolled |
Adult patients with measurable recurrent brain metastases from breast cancer.
|
||||
| Administration Time | Every 3 weeks | ||||
| Administration Dosage | 600 mg/m2 | ||||
| MOA of PDC |
Because LRP1 is also expressed on tumor cells in both CNS and systemic metastases, ANG1005 gains entry via LRP1 mediated endocytosis, where paclitaxel is cleaved from the peptide backbone by lysosomal esterases.
|
||||
| Description |
Investigator assessments resulted in median intracranial PFS of 2.8 months and the 3-month intracranial PFS rate was 52%.
|
||||
| Experiment 9 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | 3-month progression-free survival rate |
52.00%
|
|||
| Patients Enrolled |
Adult patients with measurable recurrent brain metastases from breast cancer.
|
||||
| Administration Time | Every 3 weeks | ||||
| Administration Dosage | 600 mg/m2 | ||||
| MOA of PDC |
Because LRP1 is also expressed on tumor cells in both CNS and systemic metastases, ANG1005 gains entry via LRP1 mediated endocytosis, where paclitaxel is cleaved from the peptide backbone by lysosomal esterases.
|
||||
| Description |
Investigator assessments resulted in median intracranial PFS of 2.8 months and the 3-month intracranial PFS rate was 52%.
|
||||
| Experiment 10 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | 3-month progression-free survival rate |
54.00%
|
|||
| Patients Enrolled |
Patients with leptomeningeal carcinomatosis.
|
||||
| Administration Time | Every 3 weeks | ||||
| Administration Dosage | 600 mg/m2 | ||||
| MOA of PDC |
Because LRP1 is also expressed on tumor cells in both CNS and systemic metastases, ANG1005 gains entry via LRP1 mediated endocytosis, where paclitaxel is cleaved from the peptide backbone by lysosomal esterases.
|
||||
| Description |
The investigator determined intracranial median PFS was 2.8 months and the 3-month PFS rate was 54% (Table 3). Median duration of response was 18 weeks (7.3-26.3).
|
||||
TH1902 [Phase 3]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
30%
|
|||
| Administration Time | 20 days | ||||
| Administration Dosage | 8.75 mg/kg | ||||
| MOA of PDC |
Sortilin (SORT1) is a receptor protein which cycles from the cell surface through intracellular membrane bodies. It binds several circulating proteins and peptides, including progranulin and neurotensin, prior to their rapid intracellular internalization. It is also involved in their intracellular trafficking in endosomal vesicles from the plasma membrane to lysosomes through the endocytic pathway. Deregulation in SORT1 functions has been implicated in cardiovascular disease, Alzheimers disease, and diabetes, whereas its upregulation has been documented in several forms of cancer. Recently, TH1902, a peptide-drug conjugate (PDC) to which two docetaxel molecules were linked to TH19P01 peptide, was generated. TH1902 was demonstrated to recognize and exploit SORT1 functions, and to efficiently inhibit in vitro cell proliferation, and in vivo growth of xenografts from gynecological cancers including triple-negative breast cancer (TNBC)-derived MDA-MB-231 cells, ovarian cancer-derived ES2 and SKOV3 cells, as well as endometrial cancer-derived AN3-CA cells. Interestingly, TH1902 was found to reduce in vitro vasculogenic mimicry (VM) processes in breast and ovarian cancer cells. While some molecular aspects revealing the inter-relationship existence between CSC and VM have been addressed in breast cancer, gastrointestinal cancer, and melanoma, the specific targeting of the CSC subpopulation by TH1902 remains unexplored.
Click to Show/Hide
|
||||
| Description |
In vivo assessment in tumor-bearing animal models classically complements cell cultures data for comparing cancer treatment efficacies. For this reason, hTNBCSC and hOvCSC xenografts were implanted subcutaneously into immunodeficient mice as described in the Methods section with only 1000 cells given their highly tumorigenic nature. Three days later, the animals began receiving weekly IV bolus administration of either vehicle, docetaxel, or TH1902. Docetaxel was administered at a dose (15 mg/kg/week; for 3 cycles) in accordance with the estimated maximal tolerated dose (MTD) for mice, as well as at 1/4 of the MTD (3.75 mg/kg/week). TH1902 was administered at doses (35 and 8.75 mg/kg/week) which contained quantities of docetaxel equivalent to those in the two administrations of free docetaxel. From the size of the hTNBCSC and hOvCSC tumors, it is apparent that docetaxel had little impact on xenografts growth when administered neither at its MTD, for both hTNBCSC and hOvCSC, nor at 1/4 of this dosage as reflected by the growth curves. TH1902, when administered at a dosage equivalent to docetaxel at its MTD, provided greater tumor growth inhibition than did docetaxel for both xenograft models. Furthermore, higher dosage of administered TH1902 (up to 1.5 equivalent of docetaxel MTD) did not generate significant differences in terms of hOvCSC tumor inhibition without affecting mice body weights suggesting that TH1902, even at higher doses, is better tolerated compared to docetaxel. Then, in order to statistically compare the effects of docetaxel and TH1902 on tumor growth, the tumor sizes measured at the vehicle group endpoint were compared and statistically significant differences between the tumor sizes in vehicle-treated animals found in TH1902-treated animals for hTNBCSC and hOvCSC. Mouse body weight was used as an indicator of the morbidity associated with administration of docetaxel or TH1902. Administration of docetaxel at its MTD provoked a weight loss that approached 10% after the treatments, which is often observed in xenograft models with administration of docetaxel at this level. The body weights of animals treated with an equivalent quantity or a 1.5-fold equivalent of TH1902 were maintained at a roughly constant level throughout the experiment. The animals treated with vehicle recorded a slight weight gain (~5%) over this period while animals treated with the lower dosages of docetaxel or TH1902 were similar to the animals treated with vehicle. The body weight data indicates that TH1902 appears to be better tolerated than the equivalent quantity of free docetaxel, in addition to the fact that TH1902 is more efficacious than docetaxel when administered in vivo in the murine models of CSC tested.
Click to Show/Hide
|
||||
| In Vivo Model | Nude mice hTNBCSC cells xenograft tumor model. | ||||
| In Vitro Model | Triple-negative breast cancer | Human triple-negative breast cancer stem cell (hTNBCSC) | Homo sapiens | ||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
75%
|
|||
| Administration Time | 20 days | ||||
| Administration Dosage | 35 mg/kg | ||||
| MOA of PDC |
Sortilin (SORT1) is a receptor protein which cycles from the cell surface through intracellular membrane bodies. It binds several circulating proteins and peptides, including progranulin and neurotensin, prior to their rapid intracellular internalization. It is also involved in their intracellular trafficking in endosomal vesicles from the plasma membrane to lysosomes through the endocytic pathway. Deregulation in SORT1 functions has been implicated in cardiovascular disease, Alzheimers disease, and diabetes, whereas its upregulation has been documented in several forms of cancer. Recently, TH1902, a peptide-drug conjugate (PDC) to which two docetaxel molecules were linked to TH19P01 peptide, was generated. TH1902 was demonstrated to recognize and exploit SORT1 functions, and to efficiently inhibit in vitro cell proliferation, and in vivo growth of xenografts from gynecological cancers including triple-negative breast cancer (TNBC)-derived MDA-MB-231 cells, ovarian cancer-derived ES2 and SKOV3 cells, as well as endometrial cancer-derived AN3-CA cells. Interestingly, TH1902 was found to reduce in vitro vasculogenic mimicry (VM) processes in breast and ovarian cancer cells. While some molecular aspects revealing the inter-relationship existence between CSC and VM have been addressed in breast cancer, gastrointestinal cancer, and melanoma, the specific targeting of the CSC subpopulation by TH1902 remains unexplored.
Click to Show/Hide
|
||||
| Description |
In vivo assessment in tumor-bearing animal models classically complements cell cultures data for comparing cancer treatment efficacies. For this reason, hTNBCSC and hOvCSC xenografts were implanted subcutaneously into immunodeficient mice as described in the Methods section with only 1000 cells given their highly tumorigenic nature. Three days later, the animals began receiving weekly IV bolus administration of either vehicle, docetaxel, or TH1902. Docetaxel was administered at a dose (15 mg/kg/week; for 3 cycles) in accordance with the estimated maximal tolerated dose (MTD) for mice, as well as at 1/4 of the MTD (3.75 mg/kg/week). TH1902 was administered at doses (35 and 8.75 mg/kg/week) which contained quantities of docetaxel equivalent to those in the two administrations of free docetaxel. From the size of the hTNBCSC and hOvCSC tumors, it is apparent that docetaxel had little impact on xenografts growth when administered neither at its MTD, for both hTNBCSC and hOvCSC, nor at 1/4 of this dosage as reflected by the growth curves. TH1902, when administered at a dosage equivalent to docetaxel at its MTD, provided greater tumor growth inhibition than did docetaxel for both xenograft models. Furthermore, higher dosage of administered TH1902 (up to 1.5 equivalent of docetaxel MTD) did not generate significant differences in terms of hOvCSC tumor inhibition without affecting mice body weights suggesting that TH1902, even at higher doses, is better tolerated compared to docetaxel. Then, in order to statistically compare the effects of docetaxel and TH1902 on tumor growth, the tumor sizes measured at the vehicle group endpoint were compared and statistically significant differences between the tumor sizes in vehicle-treated animals found in TH1902-treated animals for hTNBCSC and hOvCSC. Mouse body weight was used as an indicator of the morbidity associated with administration of docetaxel or TH1902. Administration of docetaxel at its MTD provoked a weight loss that approached 10% after the treatments, which is often observed in xenograft models with administration of docetaxel at this level. The body weights of animals treated with an equivalent quantity or a 1.5-fold equivalent of TH1902 were maintained at a roughly constant level throughout the experiment. The animals treated with vehicle recorded a slight weight gain (~5%) over this period while animals treated with the lower dosages of docetaxel or TH1902 were similar to the animals treated with vehicle. The body weight data indicates that TH1902 appears to be better tolerated than the equivalent quantity of free docetaxel, in addition to the fact that TH1902 is more efficacious than docetaxel when administered in vivo in the murine models of CSC tested.
Click to Show/Hide
|
||||
| In Vivo Model | Nude mice hTNBCSC cells xenograft tumor model. | ||||
| In Vitro Model | Triple-negative breast cancer | Human triple-negative breast cancer stem cell (hTNBCSC) | Homo sapiens | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
90%
|
|||
| Administration Time | 12 days | ||||
| Administration Dosage | 35 mg/kg/wk | ||||
| MOA of PDC |
The efficacy of current chemotherapeutic treatments against most solid tumors is limited by their systemic toxicity, which is partly associated with the cytotoxic properties of agents such as docetaxel or doxorubicin. To avoid or minimize adverse effects from chemotherapeutic molecules, a promising targeted approach is through peptide-drug conjugates (PDCs) that link anticancer molecules to peptides designed to interact with receptors highly expressed on cancer cells, and which can mediate the molecules rapid internalization within those cells. One such receptor is sortilin (SORT1), also known as neurotensin receptor3, a membranebound receptor that belongs to the VPS10P family of receptors. TH19P01 peptide was recently designed to target and exploit SORT1s ligand internalization function. Studies have confirmed that both TH1902 (a docetaxel-TH19P01 conjugate) and TH1904 (a doxorubicin-TH19P01 conjugate) require a SORT1-dependent mechanism of action to exert anticancer activities. In recent preclinical studies performed in immunocompromised animal models, which are unable to produce mature T-cells, TH1902 was effective against several human SORT1-positive xenograft models including triple-negative breast cancer (TNBC), ovarian cancer, and endometrial cancer.
Click to Show/Hide
|
||||
| Description |
B16-F10 melanoma syngeneic tumors were generated, with tumor sizes monitored as described in the Methods section. Tumors in xenograft-bearing, vehicle-treated mice grew at an exponential rate. Partial inhibition of tumor growth was observed after IV administration of 15 mg/kg/wk docetaxel, whereas treatment with a docetaxel-equivalent quantity of TH1902 (35 mg/kg/wk) induced tumor regression after two treatments over the period measured. Due to rapid tumor growth, only two administrations of the test articles could be performed on a weekly schedule. B16-F10 melanoma tumors from the mice treated with either vehicle, docetaxel, or TH1902 were then excised, fixed in formalin, and immunohistochemically examined.
Click to Show/Hide
|
||||
| In Vivo Model | SORT1-positive B16-F10 cells female CD1 nude mice xenograft tumor models. | ||||
| In Vitro Model | Mouse melanoma | SORT1-positive B16-F10 cell | CVCL_0159 | ||
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
45.00%
|
|||
| Administration Time | 28 days | ||||
| Administration Dosage | 8.75 mg/kg | ||||
| MOA of PDC |
TH1902 is a peptide-drug conjugate with a payload of two docetaxel molecules ester-linked to a peptide (TH19P01) designed to recognize SORT1. Studies in breast and ovarian cancer cell lines have shown that TH1902 exploited SORT1s ligand internalization functions and exerted potent antiproliferative and anti-migratory effects. Within the cell, the docetaxel molecules are released from the conjugate and can then affect polymerization of microtubules leading to aberrant mitosis and apoptosis. Intravenous administration of TH1902 to mice bearing xenografts of MDA-MB-231 breast cancer cells demonstrated a marked superiority of TH1902 over free docetaxel in preventing the growth and relapse of subcutaneous xenografts. In a previous study, SORT1 was reported to have a role in vasculogenic mimicry (VM), and TH1902 was shown to inhibit in vitro VM in these cells and in the ES-2 ovarian cancer cell line.
Click to Show/Hide
|
||||
| Description |
In the ES-2 study, mice treated with low and high doses of TH1902 significantly inhibited the growth of tumors by, respectively, 45% and 87%, whereas both docetaxel groups, at equivalent docetaxel content, produced little effect (Figure 6A and Table 1). Low doses of docetaxel and low and high doses of TH1902 were well-tolerated with slight weight gain, whereas three weekly cycles of treatments with high doses of docetaxel (the MTD for this agent in mice) produced a small weight loss when compared to initial mice weights
Click to Show/Hide
|
||||
| In Vivo Model | Female CD-1 nude mice xenograft model. | ||||
| In Vitro Model | Ewing sarcoma | ES2 cell | CVCL_AX39 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
69.00%
|
|||
| Administration Time | 28 days | ||||
| Administration Dosage | 8.75 mg/kg | ||||
| MOA of PDC |
TH1902 is a peptide-drug conjugate with a payload of two docetaxel molecules ester-linked to a peptide (TH19P01) designed to recognize SORT1. Studies in breast and ovarian cancer cell lines have shown that TH1902 exploited SORT1s ligand internalization functions and exerted potent antiproliferative and anti-migratory effects. Within the cell, the docetaxel molecules are released from the conjugate and can then affect polymerization of microtubules leading to aberrant mitosis and apoptosis. Intravenous administration of TH1902 to mice bearing xenografts of MDA-MB-231 breast cancer cells demonstrated a marked superiority of TH1902 over free docetaxel in preventing the growth and relapse of subcutaneous xenografts. In a previous study, SORT1 was reported to have a role in vasculogenic mimicry (VM), and TH1902 was shown to inhibit in vitro VM in these cells and in the ES-2 ovarian cancer cell line.
Click to Show/Hide
|
||||
| Description |
In the SKOV3 study, mice treated with low and high doses of TH1902 showed significant inhibitions of tumor growth where only high dose of docetaxel produced this effect (Figure 6B). When compared to vehicle endpoint (Day 28), low dose of TH1902 produced similar tumor growth inhibitions when compared to high dose of docetaxel (69% vs. 84%, respectively) while high dose of TH1902 induced stronger inhibition with slight regression of tumors (Table 1). Moreover, mice treated with high dose of TH1902 showed prolonged inhibitory effect (up to Day 46) where docetaxel-treated mice could not sustain this effect, which is marked by tumor regrowth (Figure 6B).
Click to Show/Hide
|
||||
| In Vivo Model | Female CD-1 nude mice xenograft model. | ||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Endometrial cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
74.00%
|
|||
| Administration Time | 28 days | ||||
| Administration Dosage | 8.75 mg/kg | ||||
| MOA of PDC |
TH1902 is a peptide-drug conjugate with a payload of two docetaxel molecules ester-linked to a peptide (TH19P01) designed to recognize SORT1. Studies in breast and ovarian cancer cell lines have shown that TH1902 exploited SORT1s ligand internalization functions and exerted potent antiproliferative and anti-migratory effects. Within the cell, the docetaxel molecules are released from the conjugate and can then affect polymerization of microtubules leading to aberrant mitosis and apoptosis. Intravenous administration of TH1902 to mice bearing xenografts of MDA-MB-231 breast cancer cells demonstrated a marked superiority of TH1902 over free docetaxel in preventing the growth and relapse of subcutaneous xenografts. In a previous study, SORT1 was reported to have a role in vasculogenic mimicry (VM), and TH1902 was shown to inhibit in vitro VM in these cells and in the ES-2 ovarian cancer cell line.
Click to Show/Hide
|
||||
| Description |
For mice bearing endometrial tumor xenografts, a similar dosage regimen to that for ovarian tumor xenografts was used as described previously for both docetaxel and TH1902. The growth of AN3-CA tumors was inhibited by both dosages of TH1902, whereas only a high dose of docetaxel could produce this effect. When compared to vehicle endpoint, low dose of TH1902 significantly inhibited the growth of AN3-CA tumors by 74%, whereas both high doses of docetaxel and TH1902 induced tumor regressions. Interestingly, a large portion of tumors (5 out of 6) within the high-dose TH1902 group were unmeasurable or remained in regression for a prolonged period (up to Day 54), whereas tumors in the equivalent docetaxel dose were unresponsive and regrew before the end of treatments. The two high doses were diminished by half for the final two treatments due to weight loss in the animals receiving high-dose docetaxel. Rapid weight loss was associated with administration of high dosage docetaxel, but this was reversed when the dosage was halved. In contrast, only mild weight loss was observed in animals administered with high-dosage TH1902.
Click to Show/Hide
|
||||
| In Vivo Model | Female CD-1 nude mice xenograft model. | ||||
| In Vitro Model | Endometrial adenocarcinoma | AN3-CA cell | CVCL_0028 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
84.00%
|
|||
| Administration Time | 28 days | ||||
| Administration Dosage | 35 mg/kg | ||||
| MOA of PDC |
TH1902 is a peptide-drug conjugate with a payload of two docetaxel molecules ester-linked to a peptide (TH19P01) designed to recognize SORT1. Studies in breast and ovarian cancer cell lines have shown that TH1902 exploited SORT1s ligand internalization functions and exerted potent antiproliferative and anti-migratory effects. Within the cell, the docetaxel molecules are released from the conjugate and can then affect polymerization of microtubules leading to aberrant mitosis and apoptosis. Intravenous administration of TH1902 to mice bearing xenografts of MDA-MB-231 breast cancer cells demonstrated a marked superiority of TH1902 over free docetaxel in preventing the growth and relapse of subcutaneous xenografts. In a previous study, SORT1 was reported to have a role in vasculogenic mimicry (VM), and TH1902 was shown to inhibit in vitro VM in these cells and in the ES-2 ovarian cancer cell line.
Click to Show/Hide
|
||||
| Description |
In the SKOV3 study, mice treated with low and high doses of TH1902 showed significant inhibitions of tumor growth where only high dose of docetaxel produced this effect (Figure 6B). When compared to vehicle endpoint (Day 28), low dose of TH1902 produced similar tumor growth inhibitions when compared to high dose of docetaxel (69% vs. 84%, respectively) while high dose of TH1902 induced stronger inhibition with slight regression of tumors (Table 1). Moreover, mice treated with high dose of TH1902 showed prolonged inhibitory effect (up to Day 46) where docetaxel-treated mice could not sustain this effect, which is marked by tumor regrowth (Figure 6B).
Click to Show/Hide
|
||||
| In Vivo Model | Female CD-1 nude mice xenograft model. | ||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
87.00%
|
|||
| Administration Time | 28 days | ||||
| Administration Dosage | 35 mg/kg | ||||
| MOA of PDC |
TH1902 is a peptide-drug conjugate with a payload of two docetaxel molecules ester-linked to a peptide (TH19P01) designed to recognize SORT1. Studies in breast and ovarian cancer cell lines have shown that TH1902 exploited SORT1s ligand internalization functions and exerted potent antiproliferative and anti-migratory effects. Within the cell, the docetaxel molecules are released from the conjugate and can then affect polymerization of microtubules leading to aberrant mitosis and apoptosis. Intravenous administration of TH1902 to mice bearing xenografts of MDA-MB-231 breast cancer cells demonstrated a marked superiority of TH1902 over free docetaxel in preventing the growth and relapse of subcutaneous xenografts. In a previous study, SORT1 was reported to have a role in vasculogenic mimicry (VM), and TH1902 was shown to inhibit in vitro VM in these cells and in the ES-2 ovarian cancer cell line.
Click to Show/Hide
|
||||
| Description |
In the ES-2 study, mice treated with low and high doses of TH1902 significantly inhibited the growth of tumors by, respectively, 45% and 87%, whereas both docetaxel groups, at equivalent docetaxel content, produced little effect (Figure 6A and Table 1). Low doses of docetaxel and low and high doses of TH1902 were well-tolerated with slight weight gain, whereas three weekly cycles of treatments with high doses of docetaxel (the MTD for this agent in mice) produced a small weight loss when compared to initial mice weights
Click to Show/Hide
|
||||
| In Vivo Model | Female CD-1 nude mice xenograft model. | ||||
| In Vitro Model | Ewing sarcoma | ES2 cell | CVCL_AX39 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Endometrial cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
100.00%
|
|||
| Administration Time | 28 days | ||||
| Administration Dosage | 35 mg/kg | ||||
| MOA of PDC |
TH1902 is a peptide-drug conjugate with a payload of two docetaxel molecules ester-linked to a peptide (TH19P01) designed to recognize SORT1. Studies in breast and ovarian cancer cell lines have shown that TH1902 exploited SORT1s ligand internalization functions and exerted potent antiproliferative and anti-migratory effects. Within the cell, the docetaxel molecules are released from the conjugate and can then affect polymerization of microtubules leading to aberrant mitosis and apoptosis. Intravenous administration of TH1902 to mice bearing xenografts of MDA-MB-231 breast cancer cells demonstrated a marked superiority of TH1902 over free docetaxel in preventing the growth and relapse of subcutaneous xenografts. In a previous study, SORT1 was reported to have a role in vasculogenic mimicry (VM), and TH1902 was shown to inhibit in vitro VM in these cells and in the ES-2 ovarian cancer cell line.
Click to Show/Hide
|
||||
| Description |
For mice bearing endometrial tumor xenografts, a similar dosage regimen to that for ovarian tumor xenografts was used as described previously for both docetaxel and TH1902. The growth of AN3-CA tumors was inhibited by both dosages of TH1902, whereas only a high dose of docetaxel could produce this effect. When compared to vehicle endpoint, low dose of TH1902 significantly inhibited the growth of AN3-CA tumors by 74%, whereas both high doses of docetaxel and TH1902 induced tumor regressions. Interestingly, a large portion of tumors (5 out of 6) within the high-dose TH1902 group were unmeasurable or remained in regression for a prolonged period (up to Day 54), whereas tumors in the equivalent docetaxel dose were unresponsive and regrew before the end of treatments. The two high doses were diminished by half for the final two treatments due to weight loss in the animals receiving high-dose docetaxel. Rapid weight loss was associated with administration of high dosage docetaxel, but this was reversed when the dosage was halved. In contrast, only mild weight loss was observed in animals administered with high-dosage TH1902.
Click to Show/Hide
|
||||
| In Vivo Model | Female CD-1 nude mice xenograft model. | ||||
| In Vitro Model | Endometrial adenocarcinoma | AN3-CA cell | CVCL_0028 | ||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
33.30%
|
|||
| Administration Time | 5 days | ||||
| Administration Dosage | 8.75 mg/kg | ||||
| MOA of PDC |
Triple-negative breast cancer (TNBC) is a heterogeneous subgroup of cancers which lacks the expression and/or amplification of targetable biomarkers (ie, estrogen receptor, progestrogen receptor, and human epidermal growth factor receptor 2), and is often associated with the worse disease-specific outcomes than other breast cancer subtypes. Here, we report that high expression of the sortilin (SORT1) receptor correlates with the decreased survival in TNBC patients, and more importantly in those bearing lymph node metastases. By exploiting SORT1 function in ligand internalization, a new anticancer treatment strategy was designed to target SORT1-positive TNBC-derived cells both in vitro and in two in vivo tumor xenografts models. A peptide (TH19P01), which requires SORT1 for internalization and to which many anticancer drugs could be conjugated, was developed. In vitro, while the TH19P01 peptide itself did not exert any antiproliferative or apoptotic effects, the docetaxel-TH19P01 conjugate (TH1902) exerted potent antiproliferative and antimigratory activities when tested on TNBC-derived MDA-MB-231 cells. TH1902 triggered faster and more potent apoptotic cell death than did unconjugated docetaxel. The apoptotic and antimigratory effects of TH1902 were both reversed by two SORT1 ligands, neurotensin and progranulin, and on siRNA-mediated silencing of SORT1. TH1902 also altered microtubule polymerization and triggered the downregulation of the anti-apoptotic Bcl-xL biomarker. In vivo, both i.p. and i.v. administrations of TH1902 led to greater tumor regression in two MDA-MB-231 and HCC-70 murine xenograft models than did docetaxel, without inducing neutropenia. Altogether, the data demonstrates the high in vivo efficacy and safety of TH1902 against TNBC through a SORT1 receptor-mediated mechanism. This property allows for selective treatment of SORT1-positive TNBC and makes TH1902 a promising avenue for personalized therapy with the potential of improving the therapeutic window of cytotoxic anticancer drugs such as docetaxel.
Click to Show/Hide
|
||||
| Description |
The use of intravenous administration of TH1902 was next investigated in the TNBC-derived MDA-MB-231 xenograft model. Mice subjected to docetaxel treatment were administered with three intravenous injections at 15 mg/kg/wk (MTD), whereas those treated with TH1902 received five injections of an equivalent dose of docetaxel at 35 mg/kg/wk. Similar to what was seen with the intraperitoneal administration protocol, a sustained decrease in tumor size was observed following intravenous administration of TH1902 until day 70, whereas a restart of tumor growth was observed at day 50 for the docetaxel-treated mice. When lower doses, equivalent to the quarter of the MTD, were used of docetaxel (3.75 mg/kg) and TH1902 (8.75 mg/kg), tumor growth as assessed by luminescence intensity was unaffected by intravenous administration of docetaxel, whereas it was significantly inhibited in the TH1902-treated group. This was further confirmed through the measurement of the tumor burden luminescence where TH1902 reduced it significantly in comparison to vehicle- or docetaxel-treated groups. Interestingly, body weight changes remained within endpoint limits in mice on intravenous administration of either docetaxel or TH1902 (data not shown). Similar conclusions were reached on testing another TNBC-derived HCC-70 xenograft model. In fact, administration of TH1902 at 8.75 mg/kg/wk led to a 93% inhibition of HCC-70 tumor growth as compared to 24% for docetaxel alone at an equivalent dose.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Half life period | 1.44 h | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
42.00%
|
|||
| Administration Time | 5 days | ||||
| Administration Dosage | 50mg /kg/wk | ||||
| MOA of PDC |
Triple-negative breast cancer (TNBC) is a heterogeneous subgroup of cancers which lacks the expression and/or amplification of targetable biomarkers (ie, estrogen receptor, progestrogen receptor, and human epidermal growth factor receptor 2), and is often associated with the worse disease-specific outcomes than other breast cancer subtypes. Here, we report that high expression of the sortilin (SORT1) receptor correlates with the decreased survival in TNBC patients, and more importantly in those bearing lymph node metastases. By exploiting SORT1 function in ligand internalization, a new anticancer treatment strategy was designed to target SORT1-positive TNBC-derived cells both in vitro and in two in vivo tumor xenografts models. A peptide (TH19P01), which requires SORT1 for internalization and to which many anticancer drugs could be conjugated, was developed. In vitro, while the TH19P01 peptide itself did not exert any antiproliferative or apoptotic effects, the docetaxel-TH19P01 conjugate (TH1902) exerted potent antiproliferative and antimigratory activities when tested on TNBC-derived MDA-MB-231 cells. TH1902 triggered faster and more potent apoptotic cell death than did unconjugated docetaxel. The apoptotic and antimigratory effects of TH1902 were both reversed by two SORT1 ligands, neurotensin and progranulin, and on siRNA-mediated silencing of SORT1. TH1902 also altered microtubule polymerization and triggered the downregulation of the anti-apoptotic Bcl-xL biomarker. In vivo, both i.p. and i.v. administrations of TH1902 led to greater tumor regression in two MDA-MB-231 and HCC-70 murine xenograft models than did docetaxel, without inducing neutropenia. Altogether, the data demonstrates the high in vivo efficacy and safety of TH1902 against TNBC through a SORT1 receptor-mediated mechanism. This property allows for selective treatment of SORT1-positive TNBC and makes TH1902 a promising avenue for personalized therapy with the potential of improving the therapeutic window of cytotoxic anticancer drugs such as docetaxel.
Click to Show/Hide
|
||||
| Description |
The in vivo efficacy of TH1902 and docetaxel against a TNBC xenograft model was next investigated in vivo. Thus, nude mice were implanted in the right flank with MDA-MB-231-luc cancer cells, and luminescence was measured to monitor tumor growth. Mice were treated with intraperitoneal injections of either docetaxel at the MTD of 15 mg/kg/wk, or with TH1902 at the maximal injectable dose of 50 mg/kg/wk, both for five cycles of treatment. Unlike the vehicle-treated control group, where the average tumor luminescence increased over time, a significant decline in luminescence intensity was observed, starting at day 5 in the TH1902-treated group. The level of luminescence intensity was also significantly lower in the free docetaxel-treated group, when compared to that in the vehicle-treated control group, but remained higher than that for the TH1902-treated group. Interestingly, tumor relapse was observed in the docetaxel-treated group beginning at day 46, whereas a sustained decrease of tumor volume was maintained in the TH1902-treated group until day 74, at which time point a complete disappearance of the tumor was achieved. Representative images of luminescence are shown for vehicle-, docetaxel-, or TH1902-treated mice at day 14 when the control group reached the tumor volume endpoint and at day 74 post-treatment at the end of the experiment. Quantification of the residual tumor burden at days 14 and 74 after docetaxel or TH1902 treatment was performed, and the analysis confirmed a much better in vivo TH1902 efficacy profile than was seen with unconjugated docetaxel. Given the lack of apoptotic or antiproliferative effects of TH19P01, no rationale supports its further assessment in vivo. As TH1902 is considered a new chemical entity, its best control condition therefore is the unconjugated docetaxel molecule itself. Body weight of mice on intraperitoneal administration of either docetaxel or TH1902 remained within endpoint limits (-20%, data not shown).
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Half life period | 1.44 h | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
50.00%
|
|||
| Administration Time | 10 days | ||||
| Administration Dosage | 8.75 mg/kg | ||||
| MOA of PDC |
Triple-negative breast cancer (TNBC) is a heterogeneous subgroup of cancers which lacks the expression and/or amplification of targetable biomarkers (ie, estrogen receptor, progestrogen receptor, and human epidermal growth factor receptor 2), and is often associated with the worse disease-specific outcomes than other breast cancer subtypes. Here, we report that high expression of the sortilin (SORT1) receptor correlates with the decreased survival in TNBC patients, and more importantly in those bearing lymph node metastases. By exploiting SORT1 function in ligand internalization, a new anticancer treatment strategy was designed to target SORT1-positive TNBC-derived cells both in vitro and in two in vivo tumor xenografts models. A peptide (TH19P01), which requires SORT1 for internalization and to which many anticancer drugs could be conjugated, was developed. In vitro, while the TH19P01 peptide itself did not exert any antiproliferative or apoptotic effects, the docetaxel-TH19P01 conjugate (TH1902) exerted potent antiproliferative and antimigratory activities when tested on TNBC-derived MDA-MB-231 cells. TH1902 triggered faster and more potent apoptotic cell death than did unconjugated docetaxel. The apoptotic and antimigratory effects of TH1902 were both reversed by two SORT1 ligands, neurotensin and progranulin, and on siRNA-mediated silencing of SORT1. TH1902 also altered microtubule polymerization and triggered the downregulation of the anti-apoptotic Bcl-xL biomarker. In vivo, both i.p. and i.v. administrations of TH1902 led to greater tumor regression in two MDA-MB-231 and HCC-70 murine xenograft models than did docetaxel, without inducing neutropenia. Altogether, the data demonstrates the high in vivo efficacy and safety of TH1902 against TNBC through a SORT1 receptor-mediated mechanism. This property allows for selective treatment of SORT1-positive TNBC and makes TH1902 a promising avenue for personalized therapy with the potential of improving the therapeutic window of cytotoxic anticancer drugs such as docetaxel.
Click to Show/Hide
|
||||
| Description |
The use of intravenous administration of TH1902 was next investigated in the TNBC-derived MDA-MB-231 xenograft model. Mice subjected to docetaxel treatment were administered with three intravenous injections at 15 mg/kg/wk (MTD), whereas those treated with TH1902 received five injections of an equivalent dose of docetaxel at 35 mg/kg/wk. Similar to what was seen with the intraperitoneal administration protocol, a sustained decrease in tumor size was observed following intravenous administration of TH1902 until day 70, whereas a restart of tumor growth was observed at day 50 for the docetaxel-treated mice. When lower doses, equivalent to the quarter of the MTD, were used of docetaxel (3.75 mg/kg) and TH1902 (8.75 mg/kg), tumor growth as assessed by luminescence intensity was unaffected by intravenous administration of docetaxel, whereas it was significantly inhibited in the TH1902-treated group. This was further confirmed through the measurement of the tumor burden luminescence where TH1902 reduced it significantly in comparison to vehicle- or docetaxel-treated groups. Interestingly, body weight changes remained within endpoint limits in mice on intravenous administration of either docetaxel or TH1902 (data not shown). Similar conclusions were reached on testing another TNBC-derived HCC-70 xenograft model. In fact, administration of TH1902 at 8.75 mg/kg/wk led to a 93% inhibition of HCC-70 tumor growth as compared to 24% for docetaxel alone at an equivalent dose.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Half life period | 1.44 h | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
55.00%
|
|||
| Administration Time | 5 days | ||||
| Administration Dosage | 35 mg/kg/wk | ||||
| MOA of PDC |
Triple-negative breast cancer (TNBC) is a heterogeneous subgroup of cancers which lacks the expression and/or amplification of targetable biomarkers (ie, estrogen receptor, progestrogen receptor, and human epidermal growth factor receptor 2), and is often associated with the worse disease-specific outcomes than other breast cancer subtypes. Here, we report that high expression of the sortilin (SORT1) receptor correlates with the decreased survival in TNBC patients, and more importantly in those bearing lymph node metastases. By exploiting SORT1 function in ligand internalization, a new anticancer treatment strategy was designed to target SORT1-positive TNBC-derived cells both in vitro and in two in vivo tumor xenografts models. A peptide (TH19P01), which requires SORT1 for internalization and to which many anticancer drugs could be conjugated, was developed. In vitro, while the TH19P01 peptide itself did not exert any antiproliferative or apoptotic effects, the docetaxel-TH19P01 conjugate (TH1902) exerted potent antiproliferative and antimigratory activities when tested on TNBC-derived MDA-MB-231 cells. TH1902 triggered faster and more potent apoptotic cell death than did unconjugated docetaxel. The apoptotic and antimigratory effects of TH1902 were both reversed by two SORT1 ligands, neurotensin and progranulin, and on siRNA-mediated silencing of SORT1. TH1902 also altered microtubule polymerization and triggered the downregulation of the anti-apoptotic Bcl-xL biomarker. In vivo, both i.p. and i.v. administrations of TH1902 led to greater tumor regression in two MDA-MB-231 and HCC-70 murine xenograft models than did docetaxel, without inducing neutropenia. Altogether, the data demonstrates the high in vivo efficacy and safety of TH1902 against TNBC through a SORT1 receptor-mediated mechanism. This property allows for selective treatment of SORT1-positive TNBC and makes TH1902 a promising avenue for personalized therapy with the potential of improving the therapeutic window of cytotoxic anticancer drugs such as docetaxel.
Click to Show/Hide
|
||||
| Description |
The use of intravenous administration of TH1902 was next investigated in the TNBC-derived MDA-MB-231 xenograft model. Mice subjected to docetaxel treatment were administered with three intravenous injections at 15 mg/kg/wk (MTD), whereas those treated with TH1902 received five injections of an equivalent dose of docetaxel at 35 mg/kg/wk. Similar to what was seen with the intraperitoneal administration protocol, a sustained decrease in tumor size was observed following intravenous administration of TH1902 until day 70, whereas a restart of tumor growth was observed at day 50 for the docetaxel-treated mice. When lower doses, equivalent to the quarter of the MTD, were used of docetaxel (3.75 mg/kg) and TH1902 (8.75 mg/kg), tumor growth as assessed by luminescence intensity was unaffected by intravenous administration of docetaxel, whereas it was significantly inhibited in the TH1902-treated group. This was further confirmed through the measurement of the tumor burden luminescence where TH1902 reduced it significantly in comparison to vehicle- or docetaxel-treated groups. Interestingly, body weight changes remained within endpoint limits in mice on intravenous administration of either docetaxel or TH1902 (data not shown). Similar conclusions were reached on testing another TNBC-derived HCC-70 xenograft model. In fact, administration of TH1902 at 8.75 mg/kg/wk led to a 93% inhibition of HCC-70 tumor growth as compared to 24% for docetaxel alone at an equivalent dose.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Half life period | 1.44 h | ||||
| Experiment 5 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
66.70%
|
|||
| Administration Time | 15 days | ||||
| Administration Dosage | 8.75 mg/kg | ||||
| MOA of PDC |
Triple-negative breast cancer (TNBC) is a heterogeneous subgroup of cancers which lacks the expression and/or amplification of targetable biomarkers (ie, estrogen receptor, progestrogen receptor, and human epidermal growth factor receptor 2), and is often associated with the worse disease-specific outcomes than other breast cancer subtypes. Here, we report that high expression of the sortilin (SORT1) receptor correlates with the decreased survival in TNBC patients, and more importantly in those bearing lymph node metastases. By exploiting SORT1 function in ligand internalization, a new anticancer treatment strategy was designed to target SORT1-positive TNBC-derived cells both in vitro and in two in vivo tumor xenografts models. A peptide (TH19P01), which requires SORT1 for internalization and to which many anticancer drugs could be conjugated, was developed. In vitro, while the TH19P01 peptide itself did not exert any antiproliferative or apoptotic effects, the docetaxel-TH19P01 conjugate (TH1902) exerted potent antiproliferative and antimigratory activities when tested on TNBC-derived MDA-MB-231 cells. TH1902 triggered faster and more potent apoptotic cell death than did unconjugated docetaxel. The apoptotic and antimigratory effects of TH1902 were both reversed by two SORT1 ligands, neurotensin and progranulin, and on siRNA-mediated silencing of SORT1. TH1902 also altered microtubule polymerization and triggered the downregulation of the anti-apoptotic Bcl-xL biomarker. In vivo, both i.p. and i.v. administrations of TH1902 led to greater tumor regression in two MDA-MB-231 and HCC-70 murine xenograft models than did docetaxel, without inducing neutropenia. Altogether, the data demonstrates the high in vivo efficacy and safety of TH1902 against TNBC through a SORT1 receptor-mediated mechanism. This property allows for selective treatment of SORT1-positive TNBC and makes TH1902 a promising avenue for personalized therapy with the potential of improving the therapeutic window of cytotoxic anticancer drugs such as docetaxel.
Click to Show/Hide
|
||||
| Description |
The use of intravenous administration of TH1902 was next investigated in the TNBC-derived MDA-MB-231 xenograft model. Mice subjected to docetaxel treatment were administered with three intravenous injections at 15 mg/kg/wk (MTD), whereas those treated with TH1902 received five injections of an equivalent dose of docetaxel at 35 mg/kg/wk. Similar to what was seen with the intraperitoneal administration protocol, a sustained decrease in tumor size was observed following intravenous administration of TH1902 until day 70, whereas a restart of tumor growth was observed at day 50 for the docetaxel-treated mice. When lower doses, equivalent to the quarter of the MTD, were used of docetaxel (3.75 mg/kg) and TH1902 (8.75 mg/kg), tumor growth as assessed by luminescence intensity was unaffected by intravenous administration of docetaxel, whereas it was significantly inhibited in the TH1902-treated group. This was further confirmed through the measurement of the tumor burden luminescence where TH1902 reduced it significantly in comparison to vehicle- or docetaxel-treated groups. Interestingly, body weight changes remained within endpoint limits in mice on intravenous administration of either docetaxel or TH1902 (data not shown). Similar conclusions were reached on testing another TNBC-derived HCC-70 xenograft model. In fact, administration of TH1902 at 8.75 mg/kg/wk led to a 93% inhibition of HCC-70 tumor growth as compared to 24% for docetaxel alone at an equivalent dose.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Half life period | 1.44 h | ||||
| Experiment 6 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
70.00%
|
|||
| Administration Time | 10 days | ||||
| Administration Dosage | 50mg /kg/wk | ||||
| MOA of PDC |
Triple-negative breast cancer (TNBC) is a heterogeneous subgroup of cancers which lacks the expression and/or amplification of targetable biomarkers (ie, estrogen receptor, progestrogen receptor, and human epidermal growth factor receptor 2), and is often associated with the worse disease-specific outcomes than other breast cancer subtypes. Here, we report that high expression of the sortilin (SORT1) receptor correlates with the decreased survival in TNBC patients, and more importantly in those bearing lymph node metastases. By exploiting SORT1 function in ligand internalization, a new anticancer treatment strategy was designed to target SORT1-positive TNBC-derived cells both in vitro and in two in vivo tumor xenografts models. A peptide (TH19P01), which requires SORT1 for internalization and to which many anticancer drugs could be conjugated, was developed. In vitro, while the TH19P01 peptide itself did not exert any antiproliferative or apoptotic effects, the docetaxel-TH19P01 conjugate (TH1902) exerted potent antiproliferative and antimigratory activities when tested on TNBC-derived MDA-MB-231 cells. TH1902 triggered faster and more potent apoptotic cell death than did unconjugated docetaxel. The apoptotic and antimigratory effects of TH1902 were both reversed by two SORT1 ligands, neurotensin and progranulin, and on siRNA-mediated silencing of SORT1. TH1902 also altered microtubule polymerization and triggered the downregulation of the anti-apoptotic Bcl-xL biomarker. In vivo, both i.p. and i.v. administrations of TH1902 led to greater tumor regression in two MDA-MB-231 and HCC-70 murine xenograft models than did docetaxel, without inducing neutropenia. Altogether, the data demonstrates the high in vivo efficacy and safety of TH1902 against TNBC through a SORT1 receptor-mediated mechanism. This property allows for selective treatment of SORT1-positive TNBC and makes TH1902 a promising avenue for personalized therapy with the potential of improving the therapeutic window of cytotoxic anticancer drugs such as docetaxel.
Click to Show/Hide
|
||||
| Description |
The in vivo efficacy of TH1902 and docetaxel against a TNBC xenograft model was next investigated in vivo. Thus, nude mice were implanted in the right flank with MDA-MB-231-luc cancer cells, and luminescence was measured to monitor tumor growth. Mice were treated with intraperitoneal injections of either docetaxel at the MTD of 15 mg/kg/wk, or with TH1902 at the maximal injectable dose of 50 mg/kg/wk, both for five cycles of treatment. Unlike the vehicle-treated control group, where the average tumor luminescence increased over time, a significant decline in luminescence intensity was observed, starting at day 5 in the TH1902-treated group. The level of luminescence intensity was also significantly lower in the free docetaxel-treated group, when compared to that in the vehicle-treated control group, but remained higher than that for the TH1902-treated group. Interestingly, tumor relapse was observed in the docetaxel-treated group beginning at day 46, whereas a sustained decrease of tumor volume was maintained in the TH1902-treated group until day 74, at which time point a complete disappearance of the tumor was achieved. Representative images of luminescence are shown for vehicle-, docetaxel-, or TH1902-treated mice at day 14 when the control group reached the tumor volume endpoint and at day 74 post-treatment at the end of the experiment. Quantification of the residual tumor burden at days 14 and 74 after docetaxel or TH1902 treatment was performed, and the analysis confirmed a much better in vivo TH1902 efficacy profile than was seen with unconjugated docetaxel. Given the lack of apoptotic or antiproliferative effects of TH19P01, no rationale supports its further assessment in vivo. As TH1902 is considered a new chemical entity, its best control condition therefore is the unconjugated docetaxel molecule itself. Body weight of mice on intraperitoneal administration of either docetaxel or TH1902 remained within endpoint limits (-20%, data not shown).
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Half life period | 1.44 h | ||||
| Experiment 7 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
78.60%
|
|||
| Administration Time | 15 days | ||||
| Administration Dosage | 35 mg/kg | ||||
| MOA of PDC |
Sortilin (SORT1) is a receptor protein which cycles from the cell surface through intracellular membrane bodies. It binds several circulating proteins and peptides, including progranulin and neurotensin, prior to their rapid intracellular internalization. It is also involved in their intracellular trafficking in endosomal vesicles from the plasma membrane to lysosomes through the endocytic pathway. Deregulation in SORT1 functions has been implicated in cardiovascular disease, Alzheimers disease, and diabetes, whereas its upregulation has been documented in several forms of cancer. Recently, TH1902, a peptide-drug conjugate (PDC) to which two docetaxel molecules were linked to TH19P01 peptide, was generated. TH1902 was demonstrated to recognize and exploit SORT1 functions, and to efficiently inhibit in vitro cell proliferation, and in vivo growth of xenografts from gynecological cancers including triple-negative breast cancer (TNBC)-derived MDA-MB-231 cells, ovarian cancer-derived ES2 and SKOV3 cells, as well as endometrial cancer-derived AN3-CA cells. Interestingly, TH1902 was found to reduce in vitro vasculogenic mimicry (VM) processes in breast and ovarian cancer cells. While some molecular aspects revealing the inter-relationship existence between CSC and VM have been addressed in breast cancer, gastrointestinal cancer, and melanoma, the specific targeting of the CSC subpopulation by TH1902 remains unexplored.
Click to Show/Hide
|
||||
| Description |
In vivo assessment in tumor-bearing animal models classically complements cell cultures data for comparing cancer treatment efficacies. For this reason, hTNBCSC and hOvCSC xenografts were implanted subcutaneously into immunodeficient mice as described in the Methods section with only 1000 cells given their highly tumorigenic nature. Three days later, the animals began receiving weekly IV bolus administration of either vehicle, docetaxel, or TH1902. Docetaxel was administered at a dose (15 mg/kg/week; for 3 cycles) in accordance with the estimated maximal tolerated dose (MTD) for mice, as well as at 1/4 of the MTD (3.75 mg/kg/week). TH1902 was administered at doses (35 and 8.75 mg/kg/week) which contained quantities of docetaxel equivalent to those in the two administrations of free docetaxel. From the size of the hTNBCSC and hOvCSC tumors, it is apparent that docetaxel had little impact on xenografts growth when administered neither at its MTD, for both hTNBCSC and hOvCSC, nor at 1/4 of this dosage as reflected by the growth curves. TH1902, when administered at a dosage equivalent to docetaxel at its MTD, provided greater tumor growth inhibition than did docetaxel for both xenograft models. Furthermore, higher dosage of administered TH1902 (up to 1.5 equivalent of docetaxel MTD) did not generate significant differences in terms of hOvCSC tumor inhibition without affecting mice body weights suggesting that TH1902, even at higher doses, is better tolerated compared to docetaxel. Then, in order to statistically compare the effects of docetaxel and TH1902 on tumor growth, the tumor sizes measured at the vehicle group endpoint were compared and statistically significant differences between the tumor sizes in vehicle-treated animals found in TH1902-treated animals for hTNBCSC and hOvCSC. Mouse body weight was used as an indicator of the morbidity associated with administration of docetaxel or TH1902. Administration of docetaxel at its MTD provoked a weight loss that approached 10% after the treatments, which is often observed in xenograft models with administration of docetaxel at this level. The body weights of animals treated with an equivalent quantity or a 1.5-fold equivalent of TH1902 were maintained at a roughly constant level throughout the experiment. The animals treated with vehicle recorded a slight weight gain (~5%) over this period while animals treated with the lower dosages of docetaxel or TH1902 were similar to the animals treated with vehicle. The body weight data indicates that TH1902 appears to be better tolerated than the equivalent quantity of free docetaxel, in addition to the fact that TH1902 is more efficacious than docetaxel when administered in vivo in the murine models of CSC tested.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian cancer | Ovarian cancer stem cell (hOvCSC) | Homo sapiens | ||
| Experiment 8 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
79%
|
|||
| Administration Time | 15 days | ||||
| Administration Dosage | 42.75 mg/kg | ||||
| MOA of PDC |
Sortilin (SORT1) is a receptor protein which cycles from the cell surface through intracellular membrane bodies. It binds several circulating proteins and peptides, including progranulin and neurotensin, prior to their rapid intracellular internalization. It is also involved in their intracellular trafficking in endosomal vesicles from the plasma membrane to lysosomes through the endocytic pathway. Deregulation in SORT1 functions has been implicated in cardiovascular disease, Alzheimers disease, and diabetes, whereas its upregulation has been documented in several forms of cancer. Recently, TH1902, a peptide-drug conjugate (PDC) to which two docetaxel molecules were linked to TH19P01 peptide, was generated. TH1902 was demonstrated to recognize and exploit SORT1 functions, and to efficiently inhibit in vitro cell proliferation, and in vivo growth of xenografts from gynecological cancers including triple-negative breast cancer (TNBC)-derived MDA-MB-231 cells, ovarian cancer-derived ES2 and SKOV3 cells, as well as endometrial cancer-derived AN3-CA cells. Interestingly, TH1902 was found to reduce in vitro vasculogenic mimicry (VM) processes in breast and ovarian cancer cells. While some molecular aspects revealing the inter-relationship existence between CSC and VM have been addressed in breast cancer, gastrointestinal cancer, and melanoma, the specific targeting of the CSC subpopulation by TH1902 remains unexplored.
Click to Show/Hide
|
||||
| Description |
In vivo assessment in tumor-bearing animal models classically complements cell cultures data for comparing cancer treatment efficacies. For this reason, hTNBCSC and hOvCSC xenografts were implanted subcutaneously into immunodeficient mice as described in the Methods section with only 1000 cells given their highly tumorigenic nature. Three days later, the animals began receiving weekly IV bolus administration of either vehicle, docetaxel, or TH1902. Docetaxel was administered at a dose (15 mg/kg/week; for 3 cycles) in accordance with the estimated maximal tolerated dose (MTD) for mice, as well as at 1/4 of the MTD (3.75 mg/kg/week). TH1902 was administered at doses (35 and 8.75 mg/kg/week) which contained quantities of docetaxel equivalent to those in the two administrations of free docetaxel. From the size of the hTNBCSC and hOvCSC tumors, it is apparent that docetaxel had little impact on xenografts growth when administered neither at its MTD, for both hTNBCSC and hOvCSC, nor at 1/4 of this dosage as reflected by the growth curves. TH1902, when administered at a dosage equivalent to docetaxel at its MTD, provided greater tumor growth inhibition than did docetaxel for both xenograft models. Furthermore, higher dosage of administered TH1902 (up to 1.5 equivalent of docetaxel MTD) did not generate significant differences in terms of hOvCSC tumor inhibition without affecting mice body weights suggesting that TH1902, even at higher doses, is better tolerated compared to docetaxel. Then, in order to statistically compare the effects of docetaxel and TH1902 on tumor growth, the tumor sizes measured at the vehicle group endpoint were compared and statistically significant differences between the tumor sizes in vehicle-treated animals found in TH1902-treated animals for hTNBCSC and hOvCSC. Mouse body weight was used as an indicator of the morbidity associated with administration of docetaxel or TH1902. Administration of docetaxel at its MTD provoked a weight loss that approached 10% after the treatments, which is often observed in xenograft models with administration of docetaxel at this level. The body weights of animals treated with an equivalent quantity or a 1.5-fold equivalent of TH1902 were maintained at a roughly constant level throughout the experiment. The animals treated with vehicle recorded a slight weight gain (~5%) over this period while animals treated with the lower dosages of docetaxel or TH1902 were similar to the animals treated with vehicle. The body weight data indicates that TH1902 appears to be better tolerated than the equivalent quantity of free docetaxel, in addition to the fact that TH1902 is more efficacious than docetaxel when administered in vivo in the murine models of CSC tested.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian cancer | Ovarian cancer stem cell (hOvCSC) | Homo sapiens | ||
| Experiment 9 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
80.00%
|
|||
| Administration Time | 15 days | ||||
| Administration Dosage | 50mg /kg/wk | ||||
| MOA of PDC |
Triple-negative breast cancer (TNBC) is a heterogeneous subgroup of cancers which lacks the expression and/or amplification of targetable biomarkers (ie, estrogen receptor, progestrogen receptor, and human epidermal growth factor receptor 2), and is often associated with the worse disease-specific outcomes than other breast cancer subtypes. Here, we report that high expression of the sortilin (SORT1) receptor correlates with the decreased survival in TNBC patients, and more importantly in those bearing lymph node metastases. By exploiting SORT1 function in ligand internalization, a new anticancer treatment strategy was designed to target SORT1-positive TNBC-derived cells both in vitro and in two in vivo tumor xenografts models. A peptide (TH19P01), which requires SORT1 for internalization and to which many anticancer drugs could be conjugated, was developed. In vitro, while the TH19P01 peptide itself did not exert any antiproliferative or apoptotic effects, the docetaxel-TH19P01 conjugate (TH1902) exerted potent antiproliferative and antimigratory activities when tested on TNBC-derived MDA-MB-231 cells. TH1902 triggered faster and more potent apoptotic cell death than did unconjugated docetaxel. The apoptotic and antimigratory effects of TH1902 were both reversed by two SORT1 ligands, neurotensin and progranulin, and on siRNA-mediated silencing of SORT1. TH1902 also altered microtubule polymerization and triggered the downregulation of the anti-apoptotic Bcl-xL biomarker. In vivo, both i.p. and i.v. administrations of TH1902 led to greater tumor regression in two MDA-MB-231 and HCC-70 murine xenograft models than did docetaxel, without inducing neutropenia. Altogether, the data demonstrates the high in vivo efficacy and safety of TH1902 against TNBC through a SORT1 receptor-mediated mechanism. This property allows for selective treatment of SORT1-positive TNBC and makes TH1902 a promising avenue for personalized therapy with the potential of improving the therapeutic window of cytotoxic anticancer drugs such as docetaxel.
Click to Show/Hide
|
||||
| Description |
The in vivo efficacy of TH1902 and docetaxel against a TNBC xenograft model was next investigated in vivo. Thus, nude mice were implanted in the right flank with MDA-MB-231-luc cancer cells, and luminescence was measured to monitor tumor growth. Mice were treated with intraperitoneal injections of either docetaxel at the MTD of 15 mg/kg/wk, or with TH1902 at the maximal injectable dose of 50 mg/kg/wk, both for five cycles of treatment. Unlike the vehicle-treated control group, where the average tumor luminescence increased over time, a significant decline in luminescence intensity was observed, starting at day 5 in the TH1902-treated group. The level of luminescence intensity was also significantly lower in the free docetaxel-treated group, when compared to that in the vehicle-treated control group, but remained higher than that for the TH1902-treated group. Interestingly, tumor relapse was observed in the docetaxel-treated group beginning at day 46, whereas a sustained decrease of tumor volume was maintained in the TH1902-treated group until day 74, at which time point a complete disappearance of the tumor was achieved. Representative images of luminescence are shown for vehicle-, docetaxel-, or TH1902-treated mice at day 14 when the control group reached the tumor volume endpoint and at day 74 post-treatment at the end of the experiment. Quantification of the residual tumor burden at days 14 and 74 after docetaxel or TH1902 treatment was performed, and the analysis confirmed a much better in vivo TH1902 efficacy profile than was seen with unconjugated docetaxel. Given the lack of apoptotic or antiproliferative effects of TH19P01, no rationale supports its further assessment in vivo. As TH1902 is considered a new chemical entity, its best control condition therefore is the unconjugated docetaxel molecule itself. Body weight of mice on intraperitoneal administration of either docetaxel or TH1902 remained within endpoint limits (-20%, data not shown).
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Half life period | 1.44 h | ||||
| Experiment 10 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
80.00%
|
|||
| Administration Time | 10 days | ||||
| Administration Dosage | 35 mg/kg/wk | ||||
| MOA of PDC |
Triple-negative breast cancer (TNBC) is a heterogeneous subgroup of cancers which lacks the expression and/or amplification of targetable biomarkers (ie, estrogen receptor, progestrogen receptor, and human epidermal growth factor receptor 2), and is often associated with the worse disease-specific outcomes than other breast cancer subtypes. Here, we report that high expression of the sortilin (SORT1) receptor correlates with the decreased survival in TNBC patients, and more importantly in those bearing lymph node metastases. By exploiting SORT1 function in ligand internalization, a new anticancer treatment strategy was designed to target SORT1-positive TNBC-derived cells both in vitro and in two in vivo tumor xenografts models. A peptide (TH19P01), which requires SORT1 for internalization and to which many anticancer drugs could be conjugated, was developed. In vitro, while the TH19P01 peptide itself did not exert any antiproliferative or apoptotic effects, the docetaxel-TH19P01 conjugate (TH1902) exerted potent antiproliferative and antimigratory activities when tested on TNBC-derived MDA-MB-231 cells. TH1902 triggered faster and more potent apoptotic cell death than did unconjugated docetaxel. The apoptotic and antimigratory effects of TH1902 were both reversed by two SORT1 ligands, neurotensin and progranulin, and on siRNA-mediated silencing of SORT1. TH1902 also altered microtubule polymerization and triggered the downregulation of the anti-apoptotic Bcl-xL biomarker. In vivo, both i.p. and i.v. administrations of TH1902 led to greater tumor regression in two MDA-MB-231 and HCC-70 murine xenograft models than did docetaxel, without inducing neutropenia. Altogether, the data demonstrates the high in vivo efficacy and safety of TH1902 against TNBC through a SORT1 receptor-mediated mechanism. This property allows for selective treatment of SORT1-positive TNBC and makes TH1902 a promising avenue for personalized therapy with the potential of improving the therapeutic window of cytotoxic anticancer drugs such as docetaxel.
Click to Show/Hide
|
||||
| Description |
The use of intravenous administration of TH1902 was next investigated in the TNBC-derived MDA-MB-231 xenograft model. Mice subjected to docetaxel treatment were administered with three intravenous injections at 15 mg/kg/wk (MTD), whereas those treated with TH1902 received five injections of an equivalent dose of docetaxel at 35 mg/kg/wk. Similar to what was seen with the intraperitoneal administration protocol, a sustained decrease in tumor size was observed following intravenous administration of TH1902 until day 70, whereas a restart of tumor growth was observed at day 50 for the docetaxel-treated mice. When lower doses, equivalent to the quarter of the MTD, were used of docetaxel (3.75 mg/kg) and TH1902 (8.75 mg/kg), tumor growth as assessed by luminescence intensity was unaffected by intravenous administration of docetaxel, whereas it was significantly inhibited in the TH1902-treated group. This was further confirmed through the measurement of the tumor burden luminescence where TH1902 reduced it significantly in comparison to vehicle- or docetaxel-treated groups. Interestingly, body weight changes remained within endpoint limits in mice on intravenous administration of either docetaxel or TH1902 (data not shown). Similar conclusions were reached on testing another TNBC-derived HCC-70 xenograft model. In fact, administration of TH1902 at 8.75 mg/kg/wk led to a 93% inhibition of HCC-70 tumor growth as compared to 24% for docetaxel alone at an equivalent dose.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Half life period | 1.44 h | ||||
| Experiment 11 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
80.00%
|
|||
| Administration Time | 20 days | ||||
| Administration Dosage | 8.75 mg/kg | ||||
| MOA of PDC |
Triple-negative breast cancer (TNBC) is a heterogeneous subgroup of cancers which lacks the expression and/or amplification of targetable biomarkers (ie, estrogen receptor, progestrogen receptor, and human epidermal growth factor receptor 2), and is often associated with the worse disease-specific outcomes than other breast cancer subtypes. Here, we report that high expression of the sortilin (SORT1) receptor correlates with the decreased survival in TNBC patients, and more importantly in those bearing lymph node metastases. By exploiting SORT1 function in ligand internalization, a new anticancer treatment strategy was designed to target SORT1-positive TNBC-derived cells both in vitro and in two in vivo tumor xenografts models. A peptide (TH19P01), which requires SORT1 for internalization and to which many anticancer drugs could be conjugated, was developed. In vitro, while the TH19P01 peptide itself did not exert any antiproliferative or apoptotic effects, the docetaxel-TH19P01 conjugate (TH1902) exerted potent antiproliferative and antimigratory activities when tested on TNBC-derived MDA-MB-231 cells. TH1902 triggered faster and more potent apoptotic cell death than did unconjugated docetaxel. The apoptotic and antimigratory effects of TH1902 were both reversed by two SORT1 ligands, neurotensin and progranulin, and on siRNA-mediated silencing of SORT1. TH1902 also altered microtubule polymerization and triggered the downregulation of the anti-apoptotic Bcl-xL biomarker. In vivo, both i.p. and i.v. administrations of TH1902 led to greater tumor regression in two MDA-MB-231 and HCC-70 murine xenograft models than did docetaxel, without inducing neutropenia. Altogether, the data demonstrates the high in vivo efficacy and safety of TH1902 against TNBC through a SORT1 receptor-mediated mechanism. This property allows for selective treatment of SORT1-positive TNBC and makes TH1902 a promising avenue for personalized therapy with the potential of improving the therapeutic window of cytotoxic anticancer drugs such as docetaxel.
Click to Show/Hide
|
||||
| Description |
The use of intravenous administration of TH1902 was next investigated in the TNBC-derived MDA-MB-231 xenograft model. Mice subjected to docetaxel treatment were administered with three intravenous injections at 15 mg/kg/wk (MTD), whereas those treated with TH1902 received five injections of an equivalent dose of docetaxel at 35 mg/kg/wk. Similar to what was seen with the intraperitoneal administration protocol, a sustained decrease in tumor size was observed following intravenous administration of TH1902 until day 70, whereas a restart of tumor growth was observed at day 50 for the docetaxel-treated mice. When lower doses, equivalent to the quarter of the MTD, were used of docetaxel (3.75 mg/kg) and TH1902 (8.75 mg/kg), tumor growth as assessed by luminescence intensity was unaffected by intravenous administration of docetaxel, whereas it was significantly inhibited in the TH1902-treated group. This was further confirmed through the measurement of the tumor burden luminescence where TH1902 reduced it significantly in comparison to vehicle- or docetaxel-treated groups. Interestingly, body weight changes remained within endpoint limits in mice on intravenous administration of either docetaxel or TH1902 (data not shown). Similar conclusions were reached on testing another TNBC-derived HCC-70 xenograft model. In fact, administration of TH1902 at 8.75 mg/kg/wk led to a 93% inhibition of HCC-70 tumor growth as compared to 24% for docetaxel alone at an equivalent dose.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Half life period | 1.44 h | ||||
| Experiment 12 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
86.00%
|
|||
| Administration Time | 15 days | ||||
| Administration Dosage | 35 mg/kg/wk | ||||
| MOA of PDC |
Triple-negative breast cancer (TNBC) is a heterogeneous subgroup of cancers which lacks the expression and/or amplification of targetable biomarkers (ie, estrogen receptor, progestrogen receptor, and human epidermal growth factor receptor 2), and is often associated with the worse disease-specific outcomes than other breast cancer subtypes. Here, we report that high expression of the sortilin (SORT1) receptor correlates with the decreased survival in TNBC patients, and more importantly in those bearing lymph node metastases. By exploiting SORT1 function in ligand internalization, a new anticancer treatment strategy was designed to target SORT1-positive TNBC-derived cells both in vitro and in two in vivo tumor xenografts models. A peptide (TH19P01), which requires SORT1 for internalization and to which many anticancer drugs could be conjugated, was developed. In vitro, while the TH19P01 peptide itself did not exert any antiproliferative or apoptotic effects, the docetaxel-TH19P01 conjugate (TH1902) exerted potent antiproliferative and antimigratory activities when tested on TNBC-derived MDA-MB-231 cells. TH1902 triggered faster and more potent apoptotic cell death than did unconjugated docetaxel. The apoptotic and antimigratory effects of TH1902 were both reversed by two SORT1 ligands, neurotensin and progranulin, and on siRNA-mediated silencing of SORT1. TH1902 also altered microtubule polymerization and triggered the downregulation of the anti-apoptotic Bcl-xL biomarker. In vivo, both i.p. and i.v. administrations of TH1902 led to greater tumor regression in two MDA-MB-231 and HCC-70 murine xenograft models than did docetaxel, without inducing neutropenia. Altogether, the data demonstrates the high in vivo efficacy and safety of TH1902 against TNBC through a SORT1 receptor-mediated mechanism. This property allows for selective treatment of SORT1-positive TNBC and makes TH1902 a promising avenue for personalized therapy with the potential of improving the therapeutic window of cytotoxic anticancer drugs such as docetaxel.
Click to Show/Hide
|
||||
| Description |
The use of intravenous administration of TH1902 was next investigated in the TNBC-derived MDA-MB-231 xenograft model. Mice subjected to docetaxel treatment were administered with three intravenous injections at 15 mg/kg/wk (MTD), whereas those treated with TH1902 received five injections of an equivalent dose of docetaxel at 35 mg/kg/wk. Similar to what was seen with the intraperitoneal administration protocol, a sustained decrease in tumor size was observed following intravenous administration of TH1902 until day 70, whereas a restart of tumor growth was observed at day 50 for the docetaxel-treated mice. When lower doses, equivalent to the quarter of the MTD, were used of docetaxel (3.75 mg/kg) and TH1902 (8.75 mg/kg), tumor growth as assessed by luminescence intensity was unaffected by intravenous administration of docetaxel, whereas it was significantly inhibited in the TH1902-treated group. This was further confirmed through the measurement of the tumor burden luminescence where TH1902 reduced it significantly in comparison to vehicle- or docetaxel-treated groups. Interestingly, body weight changes remained within endpoint limits in mice on intravenous administration of either docetaxel or TH1902 (data not shown). Similar conclusions were reached on testing another TNBC-derived HCC-70 xenograft model. In fact, administration of TH1902 at 8.75 mg/kg/wk led to a 93% inhibition of HCC-70 tumor growth as compared to 24% for docetaxel alone at an equivalent dose.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Half life period | 1.44 h | ||||
| Experiment 13 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
87%
|
|||
| Administration Time | 15 days | ||||
| Administration Dosage | 52.5 mg/kg | ||||
| MOA of PDC |
Sortilin (SORT1) is a receptor protein which cycles from the cell surface through intracellular membrane bodies. It binds several circulating proteins and peptides, including progranulin and neurotensin, prior to their rapid intracellular internalization. It is also involved in their intracellular trafficking in endosomal vesicles from the plasma membrane to lysosomes through the endocytic pathway. Deregulation in SORT1 functions has been implicated in cardiovascular disease, Alzheimers disease, and diabetes, whereas its upregulation has been documented in several forms of cancer. Recently, TH1902, a peptide-drug conjugate (PDC) to which two docetaxel molecules were linked to TH19P01 peptide, was generated. TH1902 was demonstrated to recognize and exploit SORT1 functions, and to efficiently inhibit in vitro cell proliferation, and in vivo growth of xenografts from gynecological cancers including triple-negative breast cancer (TNBC)-derived MDA-MB-231 cells, ovarian cancer-derived ES2 and SKOV3 cells, as well as endometrial cancer-derived AN3-CA cells. Interestingly, TH1902 was found to reduce in vitro vasculogenic mimicry (VM) processes in breast and ovarian cancer cells. While some molecular aspects revealing the inter-relationship existence between CSC and VM have been addressed in breast cancer, gastrointestinal cancer, and melanoma, the specific targeting of the CSC subpopulation by TH1902 remains unexplored.
Click to Show/Hide
|
||||
| Description |
In vivo assessment in tumor-bearing animal models classically complements cell cultures data for comparing cancer treatment efficacies. For this reason, hTNBCSC and hOvCSC xenografts were implanted subcutaneously into immunodeficient mice as described in the Methods section with only 1000 cells given their highly tumorigenic nature. Three days later, the animals began receiving weekly IV bolus administration of either vehicle, docetaxel, or TH1902. Docetaxel was administered at a dose (15 mg/kg/week; for 3 cycles) in accordance with the estimated maximal tolerated dose (MTD) for mice, as well as at 1/4 of the MTD (3.75 mg/kg/week). TH1902 was administered at doses (35 and 8.75 mg/kg/week) which contained quantities of docetaxel equivalent to those in the two administrations of free docetaxel. From the size of the hTNBCSC and hOvCSC tumors, it is apparent that docetaxel had little impact on xenografts growth when administered neither at its MTD, for both hTNBCSC and hOvCSC, nor at 1/4 of this dosage as reflected by the growth curves. TH1902, when administered at a dosage equivalent to docetaxel at its MTD, provided greater tumor growth inhibition than did docetaxel for both xenograft models. Furthermore, higher dosage of administered TH1902 (up to 1.5 equivalent of docetaxel MTD) did not generate significant differences in terms of hOvCSC tumor inhibition without affecting mice body weights suggesting that TH1902, even at higher doses, is better tolerated compared to docetaxel. Then, in order to statistically compare the effects of docetaxel and TH1902 on tumor growth, the tumor sizes measured at the vehicle group endpoint were compared and statistically significant differences between the tumor sizes in vehicle-treated animals found in TH1902-treated animals for hTNBCSC and hOvCSC. Mouse body weight was used as an indicator of the morbidity associated with administration of docetaxel or TH1902. Administration of docetaxel at its MTD provoked a weight loss that approached 10% after the treatments, which is often observed in xenograft models with administration of docetaxel at this level. The body weights of animals treated with an equivalent quantity or a 1.5-fold equivalent of TH1902 were maintained at a roughly constant level throughout the experiment. The animals treated with vehicle recorded a slight weight gain (~5%) over this period while animals treated with the lower dosages of docetaxel or TH1902 were similar to the animals treated with vehicle. The body weight data indicates that TH1902 appears to be better tolerated than the equivalent quantity of free docetaxel, in addition to the fact that TH1902 is more efficacious than docetaxel when administered in vivo in the murine models of CSC tested.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian cancer | Ovarian cancer stem cell (hOvCSC) | Homo sapiens | ||
| Experiment 14 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
91.00%
|
|||
| Administration Time | 20 days | ||||
| Administration Dosage | 35 mg/kg/wk | ||||
| MOA of PDC |
Triple-negative breast cancer (TNBC) is a heterogeneous subgroup of cancers which lacks the expression and/or amplification of targetable biomarkers (ie, estrogen receptor, progestrogen receptor, and human epidermal growth factor receptor 2), and is often associated with the worse disease-specific outcomes than other breast cancer subtypes. Here, we report that high expression of the sortilin (SORT1) receptor correlates with the decreased survival in TNBC patients, and more importantly in those bearing lymph node metastases. By exploiting SORT1 function in ligand internalization, a new anticancer treatment strategy was designed to target SORT1-positive TNBC-derived cells both in vitro and in two in vivo tumor xenografts models. A peptide (TH19P01), which requires SORT1 for internalization and to which many anticancer drugs could be conjugated, was developed. In vitro, while the TH19P01 peptide itself did not exert any antiproliferative or apoptotic effects, the docetaxel-TH19P01 conjugate (TH1902) exerted potent antiproliferative and antimigratory activities when tested on TNBC-derived MDA-MB-231 cells. TH1902 triggered faster and more potent apoptotic cell death than did unconjugated docetaxel. The apoptotic and antimigratory effects of TH1902 were both reversed by two SORT1 ligands, neurotensin and progranulin, and on siRNA-mediated silencing of SORT1. TH1902 also altered microtubule polymerization and triggered the downregulation of the anti-apoptotic Bcl-xL biomarker. In vivo, both i.p. and i.v. administrations of TH1902 led to greater tumor regression in two MDA-MB-231 and HCC-70 murine xenograft models than did docetaxel, without inducing neutropenia. Altogether, the data demonstrates the high in vivo efficacy and safety of TH1902 against TNBC through a SORT1 receptor-mediated mechanism. This property allows for selective treatment of SORT1-positive TNBC and makes TH1902 a promising avenue for personalized therapy with the potential of improving the therapeutic window of cytotoxic anticancer drugs such as docetaxel.
Click to Show/Hide
|
||||
| Description |
The use of intravenous administration of TH1902 was next investigated in the TNBC-derived MDA-MB-231 xenograft model. Mice subjected to docetaxel treatment were administered with three intravenous injections at 15 mg/kg/wk (MTD), whereas those treated with TH1902 received five injections of an equivalent dose of docetaxel at 35 mg/kg/wk. Similar to what was seen with the intraperitoneal administration protocol, a sustained decrease in tumor size was observed following intravenous administration of TH1902 until day 70, whereas a restart of tumor growth was observed at day 50 for the docetaxel-treated mice. When lower doses, equivalent to the quarter of the MTD, were used of docetaxel (3.75 mg/kg) and TH1902 (8.75 mg/kg), tumor growth as assessed by luminescence intensity was unaffected by intravenous administration of docetaxel, whereas it was significantly inhibited in the TH1902-treated group. This was further confirmed through the measurement of the tumor burden luminescence where TH1902 reduced it significantly in comparison to vehicle- or docetaxel-treated groups. Interestingly, body weight changes remained within endpoint limits in mice on intravenous administration of either docetaxel or TH1902 (data not shown). Similar conclusions were reached on testing another TNBC-derived HCC-70 xenograft model. In fact, administration of TH1902 at 8.75 mg/kg/wk led to a 93% inhibition of HCC-70 tumor growth as compared to 24% for docetaxel alone at an equivalent dose.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Half life period | 1.44 h | ||||
| Experiment 15 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Residual tumor burden |
2x106 RLU
|
|||
| Administration Time | 27 days | ||||
| Administration Dosage | 35 mg/kg | ||||
| MOA of PDC |
Triple-negative breast cancer (TNBC) is a heterogeneous subgroup of cancers which lacks the expression and/or amplification of targetable biomarkers (ie, estrogen receptor, progestrogen receptor, and human epidermal growth factor receptor 2), and is often associated with the worse disease-specific outcomes than other breast cancer subtypes. Here, we report that high expression of the sortilin (SORT1) receptor correlates with the decreased survival in TNBC patients, and more importantly in those bearing lymph node metastases. By exploiting SORT1 function in ligand internalization, a new anticancer treatment strategy was designed to target SORT1-positive TNBC-derived cells both in vitro and in two in vivo tumor xenografts models. A peptide (TH19P01), which requires SORT1 for internalization and to which many anticancer drugs could be conjugated, was developed. In vitro, while the TH19P01 peptide itself did not exert any antiproliferative or apoptotic effects, the docetaxel-TH19P01 conjugate (TH1902) exerted potent antiproliferative and antimigratory activities when tested on TNBC-derived MDA-MB-231 cells. TH1902 triggered faster and more potent apoptotic cell death than did unconjugated docetaxel. The apoptotic and antimigratory effects of TH1902 were both reversed by two SORT1 ligands, neurotensin and progranulin, and on siRNA-mediated silencing of SORT1. TH1902 also altered microtubule polymerization and triggered the downregulation of the anti-apoptotic Bcl-xL biomarker. In vivo, both i.p. and i.v. administrations of TH1902 led to greater tumor regression in two MDA-MB-231 and HCC-70 murine xenograft models than did docetaxel, without inducing neutropenia. Altogether, the data demonstrates the high in vivo efficacy and safety of TH1902 against TNBC through a SORT1 receptor-mediated mechanism. This property allows for selective treatment of SORT1-positive TNBC and makes TH1902 a promising avenue for personalized therapy with the potential of improving the therapeutic window of cytotoxic anticancer drugs such as docetaxel.
Click to Show/Hide
|
||||
| Description |
The use of intravenous administration of TH1902 was next investigated in the TNBC-derived MDA-MB-231 xenograft model. Mice subjected to docetaxel treatment were administered with three intravenous injections at 15 mg/kg/wk (MTD), whereas those treated with TH1902 received five injections of an equivalent dose of docetaxel at 35 mg/kg/wk. Similar to what was seen with the intraperitoneal administration protocol, a sustained decrease in tumor size was observed following intravenous administration of TH1902 until day 70, whereas a restart of tumor growth was observed at day 50 for the docetaxel-treated mice. When lower doses, equivalent to the quarter of the MTD, were used of docetaxel (3.75 mg/kg) and TH1902 (8.75 mg/kg), tumor growth as assessed by luminescence intensity was unaffected by intravenous administration of docetaxel, whereas it was significantly inhibited in the TH1902-treated group. This was further confirmed through the measurement of the tumor burden luminescence where TH1902 reduced it significantly in comparison to vehicle- or docetaxel-treated groups. Interestingly, body weight changes remained within endpoint limits in mice on intravenous administration of either docetaxel or TH1902 (data not shown). Similar conclusions were reached on testing another TNBC-derived HCC-70 xenograft model. In fact, administration of TH1902 at 8.75 mg/kg/wk led to a 93% inhibition of HCC-70 tumor growth as compared to 24% for docetaxel alone at an equivalent dose.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Half life period | 1.44 h | ||||
| Experiment 16 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Residual tumor burden |
6x106 RLU
|
|||
| Administration Time | 14 days | ||||
| Administration Dosage | 50mg /kg/wk | ||||
| MOA of PDC |
Triple-negative breast cancer (TNBC) is a heterogeneous subgroup of cancers which lacks the expression and/or amplification of targetable biomarkers (ie, estrogen receptor, progestrogen receptor, and human epidermal growth factor receptor 2), and is often associated with the worse disease-specific outcomes than other breast cancer subtypes. Here, we report that high expression of the sortilin (SORT1) receptor correlates with the decreased survival in TNBC patients, and more importantly in those bearing lymph node metastases. By exploiting SORT1 function in ligand internalization, a new anticancer treatment strategy was designed to target SORT1-positive TNBC-derived cells both in vitro and in two in vivo tumor xenografts models. A peptide (TH19P01), which requires SORT1 for internalization and to which many anticancer drugs could be conjugated, was developed. In vitro, while the TH19P01 peptide itself did not exert any antiproliferative or apoptotic effects, the docetaxel-TH19P01 conjugate (TH1902) exerted potent antiproliferative and antimigratory activities when tested on TNBC-derived MDA-MB-231 cells. TH1902 triggered faster and more potent apoptotic cell death than did unconjugated docetaxel. The apoptotic and antimigratory effects of TH1902 were both reversed by two SORT1 ligands, neurotensin and progranulin, and on siRNA-mediated silencing of SORT1. TH1902 also altered microtubule polymerization and triggered the downregulation of the anti-apoptotic Bcl-xL biomarker. In vivo, both i.p. and i.v. administrations of TH1902 led to greater tumor regression in two MDA-MB-231 and HCC-70 murine xenograft models than did docetaxel, without inducing neutropenia. Altogether, the data demonstrates the high in vivo efficacy and safety of TH1902 against TNBC through a SORT1 receptor-mediated mechanism. This property allows for selective treatment of SORT1-positive TNBC and makes TH1902 a promising avenue for personalized therapy with the potential of improving the therapeutic window of cytotoxic anticancer drugs such as docetaxel.
Click to Show/Hide
|
||||
| Description |
The use of intravenous administration of TH1902 was next investigated in the TNBC-derived MDA-MB-231 xenograft model. Mice subjected to docetaxel treatment were administered with three intravenous injections at 15 mg/kg/wk (MTD), whereas those treated with TH1902 received five injections of an equivalent dose of docetaxel at 35 mg/kg/wk. Similar to what was seen with the intraperitoneal administration protocol, a sustained decrease in tumor size was observed following intravenous administration of TH1902 until day 70, whereas a restart of tumor growth was observed at day 50 for the docetaxel-treated mice. When lower doses, equivalent to the quarter of the MTD, were used of docetaxel (3.75 mg/kg) and TH1902 (8.75 mg/kg), tumor growth as assessed by luminescence intensity was unaffected by intravenous administration of docetaxel, whereas it was significantly inhibited in the TH1902-treated group. This was further confirmed through the measurement of the tumor burden luminescence where TH1902 reduced it significantly in comparison to vehicle- or docetaxel-treated groups. Interestingly, body weight changes remained within endpoint limits in mice on intravenous administration of either docetaxel or TH1902 (data not shown). Similar conclusions were reached on testing another TNBC-derived HCC-70 xenograft model. In fact, administration of TH1902 at 8.75 mg/kg/wk led to a 93% inhibition of HCC-70 tumor growth as compared to 24% for docetaxel alone at an equivalent dose.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Half life period | 1.44 h | ||||
| Experiment 17 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
0.24 ± 0.28 nM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Triple-negative breast cancer (TNBC) is a heterogeneous subgroup of cancers which lacks the expression and/or amplification of targetable biomarkers (ie, estrogen receptor, progestrogen receptor, and human epidermal growth factor receptor 2), and is often associated with the worse disease-specific outcomes than other breast cancer subtypes. Here, we report that high expression of the sortilin (SORT1) receptor correlates with the decreased survival in TNBC patients, and more importantly in those bearing lymph node metastases. By exploiting SORT1 function in ligand internalization, a new anticancer treatment strategy was designed to target SORT1-positive TNBC-derived cells both in vitro and in two in vivo tumor xenografts models. A peptide (TH19P01), which requires SORT1 for internalization and to which many anticancer drugs could be conjugated, was developed. In vitro, while the TH19P01 peptide itself did not exert any antiproliferative or apoptotic effects, the docetaxel-TH19P01 conjugate (TH1902) exerted potent antiproliferative and antimigratory activities when tested on TNBC-derived MDA-MB-231 cells. TH1902 triggered faster and more potent apoptotic cell death than did unconjugated docetaxel. The apoptotic and antimigratory effects of TH1902 were both reversed by two SORT1 ligands, neurotensin and progranulin, and on siRNA-mediated silencing of SORT1. TH1902 also altered microtubule polymerization and triggered the downregulation of the anti-apoptotic Bcl-xL biomarker. In vivo, both i.p. and i.v. administrations of TH1902 led to greater tumor regression in two MDA-MB-231 and HCC-70 murine xenograft models than did docetaxel, without inducing neutropenia. Altogether, the data demonstrates the high in vivo efficacy and safety of TH1902 against TNBC through a SORT1 receptor-mediated mechanism. This property allows for selective treatment of SORT1-positive TNBC and makes TH1902 a promising avenue for personalized therapy with the potential of improving the therapeutic window of cytotoxic anticancer drugs such as docetaxel.
Click to Show/Hide
|
||||
| Description |
To determine whether docetaxel or TH1902 inhibited TNBC cell proliferation, MTT assays were performed in MDA-MB-231 and HCC-70 cells exposed to various concentrations of TH19P01, docetaxel or TH1902. Inhibition of MDA-MB-231 cell proliferation was effectively triggered by both docetaxel and TH1902 compounds in a concentration-dependent manner whereas TH19P01 had no effect at the concentrations tested. The IC50 value of TH1902 was found to be comparable to that of docetaxel at low nM concentrations in both MDA-MB-231 and HCC-70 cells, which supports the rationale that conjugated docetaxel can indeed be released from TH1902 once internalized and exert its antiproliferative effect inside the targeted cancer cells. The effect of TH1902 on MBA-MB-231 cell-cycle arrest was also tested. The results show that more than 70% of treated cells were arrested in the G2/M phase of the cell cycle in comparison to vehicle-treated control cells where ~14% of the cells remained in the G2/M phase. While TH19P01 appears to be an inert peptide with no in vitro antiproliferative effects, these data confirm both the growth inhibitory effect of TH1902 and, more importantly, that the docetaxel anticancer potency was unaffected by its conjugation to the TH19P01 peptide.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Half life period | 1.44 h | ||||
| Experiment 18 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
0.29 ± 0.24 nM
|
|||
| Administration Time | 72 h | ||||
| MOA of PDC |
TH1902 is a peptide-drug conjugate with a payload of two docetaxel molecules ester-linked to a peptide (TH19P01) designed to recognize SORT1. Studies in breast and ovarian cancer cell lines have shown that TH1902 exploited SORT1s ligand internalization functions and exerted potent antiproliferative and anti-migratory effects. Within the cell, the docetaxel molecules are released from the conjugate and can then affect polymerization of microtubules leading to aberrant mitosis and apoptosis. Intravenous administration of TH1902 to mice bearing xenografts of MDA-MB-231 breast cancer cells demonstrated a marked superiority of TH1902 over free docetaxel in preventing the growth and relapse of subcutaneous xenografts. In a previous study, SORT1 was reported to have a role in vasculogenic mimicry (VM), and TH1902 was shown to inhibit in vitro VM in these cells and in the ES-2 ovarian cancer cell line.
Click to Show/Hide
|
||||
| Description |
Cells were thus exposed to various concentrations of TH1902 or docetaxel, and the effects on cell proliferation were measured using the MTT detection assay. It is apparent that both reagents exerted a cytotoxic effect with IC50 values of TH1902 comparable to that of docetaxel at low nM concentrations in the four cell lines tested. This supports the rationale that conjugated docetaxel can be released from TH1902 and exert its anti-proliferative effect inside the targeted cancer cells. Parallel experiment showed that cell proliferation was unaffected by the free peptide (TH19P01) at concentrations up to 1000 nM.
Click to Show/Hide
|
||||
| In Vitro Model | Endometrial adenocarcinoma | AN3-CA cell | CVCL_0028 | ||
| Experiment 19 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
0.38 nM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The cancer phenotype is commonly associated with aberrant glycosylation patterns. One glycan that is directly linked to cancer is the Thomsen-Friedenreich antigen (TF or CD176). It is a disaccharide composed of a galactose β1-3 N-acetylgalactosamine, O-linked to a glycoprotein through serine or threonine residues and commonly written as Galβ1-3GalNAc--O-Ser/Thr. The TF is therapeutically attractive due to its cryptic nature in normal cells and exposure in embryonic and cancer cells. The expression of the TF has been demonstrated in 90% of primary human carcinomas, including in the lung, the breast, and the pancreas. Additionally, cancer initiating cells or cancer stem cells in the lung, liver, and colon express the TF. The peptide sequence HGRFILPWWYAFSPS (TF-peptide) is known to bind tightly to the TF (Kd = 1.2 M) and has been demonstrated to inhibit processes directly involved in TF accessibility.
Click to Show/Hide
|
||||
| Description |
SORT1-positive SK-MEL-28 and B16-F10 melanoma cells were selected for testing the anti-proliferative effect of docetaxel and TH1902. When TH1902 biological effects were monitored, the half-maximal inhibitory concentration (IC50) of TH1902 was similar to that of docetaxel, averaging 0.38 vs. 0.39 nM, respectively, in human SK-MEL-28 cells, and 2.57 vs. 1.72 nM in murine B16-F10 cells.
Click to Show/Hide
|
||||
| In Vitro Model | Cutaneous melanoma | SORT1-positive SKMEL28 cell | CVCL_0526 | ||
| Experiment 20 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
0.55 ± 0.21 nM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
TH1902 is a peptide-drug conjugate with a payload of two docetaxel molecules ester-linked to a peptide (TH19P01) designed to recognize SORT1. Studies in breast and ovarian cancer cell lines have shown that TH1902 exploited SORT1s ligand internalization functions and exerted potent antiproliferative and anti-migratory effects. Within the cell, the docetaxel molecules are released from the conjugate and can then affect polymerization of microtubules leading to aberrant mitosis and apoptosis. Intravenous administration of TH1902 to mice bearing xenografts of MDA-MB-231 breast cancer cells demonstrated a marked superiority of TH1902 over free docetaxel in preventing the growth and relapse of subcutaneous xenografts. In a previous study, SORT1 was reported to have a role in vasculogenic mimicry (VM), and TH1902 was shown to inhibit in vitro VM in these cells and in the ES-2 ovarian cancer cell line.
Click to Show/Hide
|
||||
| Description |
Cells were thus exposed to various concentrations of TH1902 or docetaxel, and the effects on cell proliferation were measured using the MTT detection assay. It is apparent that both reagents exerted a cytotoxic effect with IC50 values of TH1902 comparable to that of docetaxel at low nM concentrations in the four cell lines tested. This supports the rationale that conjugated docetaxel can be released from TH1902 and exert its anti-proliferative effect inside the targeted cancer cells. Parallel experiment showed that cell proliferation was unaffected by the free peptide (TH19P01) at concentrations up to 1000 nM.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 21 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
1.00 ±0.55 nM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Triple-negative breast cancer (TNBC) is a heterogeneous subgroup of cancers which lacks the expression and/or amplification of targetable biomarkers (ie, estrogen receptor, progestrogen receptor, and human epidermal growth factor receptor 2), and is often associated with the worse disease-specific outcomes than other breast cancer subtypes. Here, we report that high expression of the sortilin (SORT1) receptor correlates with the decreased survival in TNBC patients, and more importantly in those bearing lymph node metastases. By exploiting SORT1 function in ligand internalization, a new anticancer treatment strategy was designed to target SORT1-positive TNBC-derived cells both in vitro and in two in vivo tumor xenografts models. A peptide (TH19P01), which requires SORT1 for internalization and to which many anticancer drugs could be conjugated, was developed. In vitro, while the TH19P01 peptide itself did not exert any antiproliferative or apoptotic effects, the docetaxel-TH19P01 conjugate (TH1902) exerted potent antiproliferative and antimigratory activities when tested on TNBC-derived MDA-MB-231 cells. TH1902 triggered faster and more potent apoptotic cell death than did unconjugated docetaxel. The apoptotic and antimigratory effects of TH1902 were both reversed by two SORT1 ligands, neurotensin and progranulin, and on siRNA-mediated silencing of SORT1. TH1902 also altered microtubule polymerization and triggered the downregulation of the anti-apoptotic Bcl-xL biomarker. In vivo, both i.p. and i.v. administrations of TH1902 led to greater tumor regression in two MDA-MB-231 and HCC-70 murine xenograft models than did docetaxel, without inducing neutropenia. Altogether, the data demonstrates the high in vivo efficacy and safety of TH1902 against TNBC through a SORT1 receptor-mediated mechanism. This property allows for selective treatment of SORT1-positive TNBC and makes TH1902 a promising avenue for personalized therapy with the potential of improving the therapeutic window of cytotoxic anticancer drugs such as docetaxel.
Click to Show/Hide
|
||||
| Description |
To determine whether docetaxel or TH1902 inhibited TNBC cell proliferation, MTT assays were performed in MDA-MB-231 and HCC-70 cells exposed to various concentrations of TH19P01, docetaxel or TH1902. Inhibition of MDA-MB-231 cell proliferation was effectively triggered by both docetaxel and TH1902 compounds in a concentration-dependent manner whereas TH19P01 had no effect at the concentrations tested. The IC50 value of TH1902 was found to be comparable to that of docetaxel at low nM concentrations in both MDA-MB-231 and HCC-70 cells, which supports the rationale that conjugated docetaxel can indeed be released from TH1902 once internalized and exert its antiproliferative effect inside the targeted cancer cells. The effect of TH1902 on MBA-MB-231 cell-cycle arrest was also tested. The results show that more than 70% of treated cells were arrested in the G2/M phase of the cell cycle in comparison to vehicle-treated control cells where ~14% of the cells remained in the G2/M phase. While TH19P01 appears to be an inert peptide with no in vitro antiproliferative effects, these data confirm both the growth inhibitory effect of TH1902 and, more importantly, that the docetaxel anticancer potency was unaffected by its conjugation to the TH19P01 peptide.
Click to Show/Hide
|
||||
| In Vitro Model | Breast ductal carcinoma | HCC70 cell | CVCL_1270 | ||
| Half life period | 1.44 h | ||||
| Experiment 22 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
1.58±0.07 nM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
TH1902 is a peptide-drug conjugate with a payload of two docetaxel molecules ester-linked to a peptide (TH19P01) designed to recognize SORT1. Studies in breast and ovarian cancer cell lines have shown that TH1902 exploited SORT1s ligand internalization functions and exerted potent antiproliferative and anti-migratory effects. Within the cell, the docetaxel molecules are released from the conjugate and can then affect polymerization of microtubules leading to aberrant mitosis and apoptosis. Intravenous administration of TH1902 to mice bearing xenografts of MDA-MB-231 breast cancer cells demonstrated a marked superiority of TH1902 over free docetaxel in preventing the growth and relapse of subcutaneous xenografts. In a previous study, SORT1 was reported to have a role in vasculogenic mimicry (VM), and TH1902 was shown to inhibit in vitro VM in these cells and in the ES-2 ovarian cancer cell line.
Click to Show/Hide
|
||||
| Description |
For mice bearing endometrial tumor xenografts, a similar dosage regimen to that for ovarian tumor xenografts was used as described previously for both docetaxel and TH1902. The growth of AN3-CA tumors was inhibited by both dosages of TH1902, whereas only
|
||||
| In Vitro Model | Ovarian clear cell adenocarcinoma | ES-2 cell | CVCL_3509 | ||
| Experiment 23 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.57 nM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
As a payload for PDCs (and ADCs), analogues of the duocarmycins are attractive. Duocarmycin SA and yatakemycin rank among the most potent natural cytotoxins discovered. The cyclopropyl and prodrug seco forms are both naturally occurring and equipotent in most circumstances. Studies of the binding-driven bonding model of their interaction with DNA suggest that their utility will be enhanced when targeted to tumor cells. In fact, SYD985, an ADC that utilizes a peptide linker for a duocarmycin analogue to trastuzumab, has recently been progressed to phase III clinical trial.
Click to Show/Hide
|
||||
| Description |
SORT1-positive SK-MEL-28 and B16-F10 melanoma cells were selected for testing the anti-proliferative effect of docetaxel and TH1902. When TH1902 biological effects were monitored, the half-maximal inhibitory concentration (IC50) of TH1902 was similar to that of docetaxel, averaging 0.38 vs. 0.39 nM, respectively, in human SK-MEL-28 cells, and 2.57 vs. 1.72 nM in murine B16-F10 cells.
Click to Show/Hide
|
||||
| In Vitro Model | Mouse melanoma | SORT1-positive B16-F10 cell | CVCL_0159 | ||
| Experiment 24 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Endometrial cancer | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
3.99 ± 0.96 nM
|
|||
| Administration Time | 72 h | ||||
| MOA of PDC |
TH1902 is a peptide-drug conjugate with a payload of two docetaxel molecules ester-linked to a peptide (TH19P01) designed to recognize SORT1. Studies in breast and ovarian cancer cell lines have shown that TH1902 exploited SORT1s ligand internalization functions and exerted potent antiproliferative and anti-migratory effects. Within the cell, the docetaxel molecules are released from the conjugate and can then affect polymerization of microtubules leading to aberrant mitosis and apoptosis. Intravenous administration of TH1902 to mice bearing xenografts of MDA-MB-231 breast cancer cells demonstrated a marked superiority of TH1902 over free docetaxel in preventing the growth and relapse of subcutaneous xenografts. In a previous study, SORT1 was reported to have a role in vasculogenic mimicry (VM), and TH1902 was shown to inhibit in vitro VM in these cells and in the ES-2 ovarian cancer cell line.
Click to Show/Hide
|
||||
| Description |
Cells were thus exposed to various concentrations of TH1902 or docetaxel, and the effects on cell proliferation were measured using the MTT detection assay. It is apparent that both reagents exerted a cytotoxic effect with IC50 values of TH1902 comparable to that of docetaxel at low nM concentrations in the four cell lines tested. This supports the rationale that conjugated docetaxel can be released from TH1902 and exert its anti-proliferative effect inside the targeted cancer cells. Parallel experiment showed that cell proliferation was unaffected by the free peptide (TH19P01) at concentrations up to 1000 nM.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 25 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Apotosis rate |
9.00%
|
|||
| Administration Time | 5 h | ||||
| Administration Dosage | 5 μM | ||||
| Evaluation Method | Flow cytometry | ||||
| MOA of PDC |
Triple-negative breast cancer (TNBC) is a heterogeneous subgroup of cancers which lacks the expression and/or amplification of targetable biomarkers (ie, estrogen receptor, progestrogen receptor, and human epidermal growth factor receptor 2), and is often associated with the worse disease-specific outcomes than other breast cancer subtypes. Here, we report that high expression of the sortilin (SORT1) receptor correlates with the decreased survival in TNBC patients, and more importantly in those bearing lymph node metastases. By exploiting SORT1 function in ligand internalization, a new anticancer treatment strategy was designed to target SORT1-positive TNBC-derived cells both in vitro and in two in vivo tumor xenografts models. A peptide (TH19P01), which requires SORT1 for internalization and to which many anticancer drugs could be conjugated, was developed. In vitro, while the TH19P01 peptide itself did not exert any antiproliferative or apoptotic effects, the docetaxel-TH19P01 conjugate (TH1902) exerted potent antiproliferative and antimigratory activities when tested on TNBC-derived MDA-MB-231 cells. TH1902 triggered faster and more potent apoptotic cell death than did unconjugated docetaxel. The apoptotic and antimigratory effects of TH1902 were both reversed by two SORT1 ligands, neurotensin and progranulin, and on siRNA-mediated silencing of SORT1. TH1902 also altered microtubule polymerization and triggered the downregulation of the anti-apoptotic Bcl-xL biomarker. In vivo, both i.p. and i.v. administrations of TH1902 led to greater tumor regression in two MDA-MB-231 and HCC-70 murine xenograft models than did docetaxel, without inducing neutropenia. Altogether, the data demonstrates the high in vivo efficacy and safety of TH1902 against TNBC through a SORT1 receptor-mediated mechanism. This property allows for selective treatment of SORT1-positive TNBC and makes TH1902 a promising avenue for personalized therapy with the potential of improving the therapeutic window of cytotoxic anticancer drugs such as docetaxel.
Click to Show/Hide
|
||||
| Description |
The impact of docetaxel and TH1902 on MDA-MB-231 cellular morphology was evaluated and visualized by light microscopy. A drastic change in cell shape was observed in both docetaxel- and TH1902-treated cells when compared to control cells. As already reported for docetaxel-treated human renal clear cell carcinoma, MDA-MB-231 cells treated with docetaxel appeared more contracted than control cells. Interestingly, TH1902-treated cell morphology appeared even smaller, less flattened, and presented greater inter-cell spacing in comparison to those treated with free docetaxel. This observation confirms the enhanced cytotoxic effect of TH1902 when compared to docetaxel. Docetaxel is further believed to exert its cytotoxic effects through both cell cycle regulation and apoptosis induction. MDA-MB-231 cells were therefore treated for 5 or 24 hours with various concentrations of either docetaxel or TH1902, followed by AnnexinV/PI staining. TH1902 increased MDA-MB-231 cell apoptosis in a dose-dependent manner when compared to docetaxel treatment. Less than 1% apoptotic cells were measured when cells were treated with 50 μM TH19P01 for 24 hours (data not shown). Overall, this suggests that, even within such a short time frame, receptor-mediated events account for the increased effects of TH1902. Finally, to confirm the role of SORT1 in TH1902 internalization and induced apoptosis, pharmacological inhibition of its apoptotic activity was measured after incubating MDA-MB-231 breast cancer cells with TH1902 in the presence or absence of the free unlabeled TH19P01 peptide or of one of the two different SORT1 ligands neurotensin or progranulin. The apoptosis assay showed that excess TH19P01 peptide, neurotensin or progranulin competitively reduced TH1902-induced cell death by 68%, 53% and 87%, respectively. This confirms the involvement of SORT1 in the internalization process of TH1902 leading to cell death in a TNBC cell model.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Half life period | 1.44 h | ||||
| Experiment 26 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Apotosis rate |
19.00%
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 5 μM | ||||
| Evaluation Method | Flow cytometry | ||||
| MOA of PDC |
Triple-negative breast cancer (TNBC) is a heterogeneous subgroup of cancers which lacks the expression and/or amplification of targetable biomarkers (ie, estrogen receptor, progestrogen receptor, and human epidermal growth factor receptor 2), and is often associated with the worse disease-specific outcomes than other breast cancer subtypes. Here, we report that high expression of the sortilin (SORT1) receptor correlates with the decreased survival in TNBC patients, and more importantly in those bearing lymph node metastases. By exploiting SORT1 function in ligand internalization, a new anticancer treatment strategy was designed to target SORT1-positive TNBC-derived cells both in vitro and in two in vivo tumor xenografts models. A peptide (TH19P01), which requires SORT1 for internalization and to which many anticancer drugs could be conjugated, was developed. In vitro, while the TH19P01 peptide itself did not exert any antiproliferative or apoptotic effects, the docetaxel-TH19P01 conjugate (TH1902) exerted potent antiproliferative and antimigratory activities when tested on TNBC-derived MDA-MB-231 cells. TH1902 triggered faster and more potent apoptotic cell death than did unconjugated docetaxel. The apoptotic and antimigratory effects of TH1902 were both reversed by two SORT1 ligands, neurotensin and progranulin, and on siRNA-mediated silencing of SORT1. TH1902 also altered microtubule polymerization and triggered the downregulation of the anti-apoptotic Bcl-xL biomarker. In vivo, both i.p. and i.v. administrations of TH1902 led to greater tumor regression in two MDA-MB-231 and HCC-70 murine xenograft models than did docetaxel, without inducing neutropenia. Altogether, the data demonstrates the high in vivo efficacy and safety of TH1902 against TNBC through a SORT1 receptor-mediated mechanism. This property allows for selective treatment of SORT1-positive TNBC and makes TH1902 a promising avenue for personalized therapy with the potential of improving the therapeutic window of cytotoxic anticancer drugs such as docetaxel.
Click to Show/Hide
|
||||
| Description |
The impact of docetaxel and TH1902 on MDA-MB-231 cellular morphology was evaluated and visualized by light microscopy. A drastic change in cell shape was observed in both docetaxel- and TH1902-treated cells when compared to control cells. As already reported for docetaxel-treated human renal clear cell carcinoma, MDA-MB-231 cells treated with docetaxel appeared more contracted than control cells. Interestingly, TH1902-treated cell morphology appeared even smaller, less flattened, and presented greater inter-cell spacing in comparison to those treated with free docetaxel. This observation confirms the enhanced cytotoxic effect of TH1902 when compared to docetaxel. Docetaxel is further believed to exert its cytotoxic effects through both cell cycle regulation and apoptosis induction. MDA-MB-231 cells were therefore treated for 5 or 24 hours with various concentrations of either docetaxel or TH1902, followed by AnnexinV/PI staining. TH1902 increased MDA-MB-231 cell apoptosis in a dose-dependent manner when compared to docetaxel treatment. Less than 1% apoptotic cells were measured when cells were treated with 50 μM TH19P01 for 24 hours (data not shown). Overall, this suggests that, even within such a short time frame, receptor-mediated events account for the increased effects of TH1902. Finally, to confirm the role of SORT1 in TH1902 internalization and induced apoptosis, pharmacological inhibition of its apoptotic activity was measured after incubating MDA-MB-231 breast cancer cells with TH1902 in the presence or absence of the free unlabeled TH19P01 peptide or of one of the two different SORT1 ligands neurotensin or progranulin. The apoptosis assay showed that excess TH19P01 peptide, neurotensin or progranulin competitively reduced TH1902-induced cell death by 68%, 53% and 87%, respectively. This confirms the involvement of SORT1 in the internalization process of TH1902 leading to cell death in a TNBC cell model.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Half life period | 1.44 h | ||||
| Experiment 27 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Apotosis rate |
22.00%
|
|||
| Administration Time | 5 h | ||||
| Administration Dosage | 10 μM | ||||
| Evaluation Method | Flow cytometry | ||||
| MOA of PDC |
Triple-negative breast cancer (TNBC) is a heterogeneous subgroup of cancers which lacks the expression and/or amplification of targetable biomarkers (ie, estrogen receptor, progestrogen receptor, and human epidermal growth factor receptor 2), and is often associated with the worse disease-specific outcomes than other breast cancer subtypes. Here, we report that high expression of the sortilin (SORT1) receptor correlates with the decreased survival in TNBC patients, and more importantly in those bearing lymph node metastases. By exploiting SORT1 function in ligand internalization, a new anticancer treatment strategy was designed to target SORT1-positive TNBC-derived cells both in vitro and in two in vivo tumor xenografts models. A peptide (TH19P01), which requires SORT1 for internalization and to which many anticancer drugs could be conjugated, was developed. In vitro, while the TH19P01 peptide itself did not exert any antiproliferative or apoptotic effects, the docetaxel-TH19P01 conjugate (TH1902) exerted potent antiproliferative and antimigratory activities when tested on TNBC-derived MDA-MB-231 cells. TH1902 triggered faster and more potent apoptotic cell death than did unconjugated docetaxel. The apoptotic and antimigratory effects of TH1902 were both reversed by two SORT1 ligands, neurotensin and progranulin, and on siRNA-mediated silencing of SORT1. TH1902 also altered microtubule polymerization and triggered the downregulation of the anti-apoptotic Bcl-xL biomarker. In vivo, both i.p. and i.v. administrations of TH1902 led to greater tumor regression in two MDA-MB-231 and HCC-70 murine xenograft models than did docetaxel, without inducing neutropenia. Altogether, the data demonstrates the high in vivo efficacy and safety of TH1902 against TNBC through a SORT1 receptor-mediated mechanism. This property allows for selective treatment of SORT1-positive TNBC and makes TH1902 a promising avenue for personalized therapy with the potential of improving the therapeutic window of cytotoxic anticancer drugs such as docetaxel.
Click to Show/Hide
|
||||
| Description |
The impact of docetaxel and TH1902 on MDA-MB-231 cellular morphology was evaluated and visualized by light microscopy. A drastic change in cell shape was observed in both docetaxel- and TH1902-treated cells when compared to control cells. As already reported for docetaxel-treated human renal clear cell carcinoma, MDA-MB-231 cells treated with docetaxel appeared more contracted than control cells. Interestingly, TH1902-treated cell morphology appeared even smaller, less flattened, and presented greater inter-cell spacing in comparison to those treated with free docetaxel. This observation confirms the enhanced cytotoxic effect of TH1902 when compared to docetaxel. Docetaxel is further believed to exert its cytotoxic effects through both cell cycle regulation and apoptosis induction. MDA-MB-231 cells were therefore treated for 5 or 24 hours with various concentrations of either docetaxel or TH1902, followed by AnnexinV/PI staining. TH1902 increased MDA-MB-231 cell apoptosis in a dose-dependent manner when compared to docetaxel treatment. Less than 1% apoptotic cells were measured when cells were treated with 50 μM TH19P01 for 24 hours (data not shown). Overall, this suggests that, even within such a short time frame, receptor-mediated events account for the increased effects of TH1902. Finally, to confirm the role of SORT1 in TH1902 internalization and induced apoptosis, pharmacological inhibition of its apoptotic activity was measured after incubating MDA-MB-231 breast cancer cells with TH1902 in the presence or absence of the free unlabeled TH19P01 peptide or of one of the two different SORT1 ligands neurotensin or progranulin. The apoptosis assay showed that excess TH19P01 peptide, neurotensin or progranulin competitively reduced TH1902-induced cell death by 68%, 53% and 87%, respectively. This confirms the involvement of SORT1 in the internalization process of TH1902 leading to cell death in a TNBC cell model.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Half life period | 1.44 h | ||||
| Experiment 28 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Apotosis rate |
35.00%
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 10 μM | ||||
| Evaluation Method | Flow cytometry | ||||
| MOA of PDC |
Triple-negative breast cancer (TNBC) is a heterogeneous subgroup of cancers which lacks the expression and/or amplification of targetable biomarkers (ie, estrogen receptor, progestrogen receptor, and human epidermal growth factor receptor 2), and is often associated with the worse disease-specific outcomes than other breast cancer subtypes. Here, we report that high expression of the sortilin (SORT1) receptor correlates with the decreased survival in TNBC patients, and more importantly in those bearing lymph node metastases. By exploiting SORT1 function in ligand internalization, a new anticancer treatment strategy was designed to target SORT1-positive TNBC-derived cells both in vitro and in two in vivo tumor xenografts models. A peptide (TH19P01), which requires SORT1 for internalization and to which many anticancer drugs could be conjugated, was developed. In vitro, while the TH19P01 peptide itself did not exert any antiproliferative or apoptotic effects, the docetaxel-TH19P01 conjugate (TH1902) exerted potent antiproliferative and antimigratory activities when tested on TNBC-derived MDA-MB-231 cells. TH1902 triggered faster and more potent apoptotic cell death than did unconjugated docetaxel. The apoptotic and antimigratory effects of TH1902 were both reversed by two SORT1 ligands, neurotensin and progranulin, and on siRNA-mediated silencing of SORT1. TH1902 also altered microtubule polymerization and triggered the downregulation of the anti-apoptotic Bcl-xL biomarker. In vivo, both i.p. and i.v. administrations of TH1902 led to greater tumor regression in two MDA-MB-231 and HCC-70 murine xenograft models than did docetaxel, without inducing neutropenia. Altogether, the data demonstrates the high in vivo efficacy and safety of TH1902 against TNBC through a SORT1 receptor-mediated mechanism. This property allows for selective treatment of SORT1-positive TNBC and makes TH1902 a promising avenue for personalized therapy with the potential of improving the therapeutic window of cytotoxic anticancer drugs such as docetaxel.
Click to Show/Hide
|
||||
| Description |
The impact of docetaxel and TH1902 on MDA-MB-231 cellular morphology was evaluated and visualized by light microscopy. A drastic change in cell shape was observed in both docetaxel- and TH1902-treated cells when compared to control cells. As already reported for docetaxel-treated human renal clear cell carcinoma, MDA-MB-231 cells treated with docetaxel appeared more contracted than control cells. Interestingly, TH1902-treated cell morphology appeared even smaller, less flattened, and presented greater inter-cell spacing in comparison to those treated with free docetaxel. This observation confirms the enhanced cytotoxic effect of TH1902 when compared to docetaxel. Docetaxel is further believed to exert its cytotoxic effects through both cell cycle regulation and apoptosis induction. MDA-MB-231 cells were therefore treated for 5 or 24 hours with various concentrations of either docetaxel or TH1902, followed by AnnexinV/PI staining. TH1902 increased MDA-MB-231 cell apoptosis in a dose-dependent manner when compared to docetaxel treatment. Less than 1% apoptotic cells were measured when cells were treated with 50 μM TH19P01 for 24 hours (data not shown). Overall, this suggests that, even within such a short time frame, receptor-mediated events account for the increased effects of TH1902. Finally, to confirm the role of SORT1 in TH1902 internalization and induced apoptosis, pharmacological inhibition of its apoptotic activity was measured after incubating MDA-MB-231 breast cancer cells with TH1902 in the presence or absence of the free unlabeled TH19P01 peptide or of one of the two different SORT1 ligands neurotensin or progranulin. The apoptosis assay showed that excess TH19P01 peptide, neurotensin or progranulin competitively reduced TH1902-induced cell death by 68%, 53% and 87%, respectively. This confirms the involvement of SORT1 in the internalization process of TH1902 leading to cell death in a TNBC cell model.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Half life period | 1.44 h | ||||
| Experiment 29 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Apotosis rate |
45.00%
|
|||
| Administration Time | 5 h | ||||
| Administration Dosage | 2 μM | ||||
| Evaluation Method | Annexin X/PI staining combined with flow cytometry | ||||
| MOA of PDC |
TH1902 is a peptide-drug conjugate with a payload of two docetaxel molecules ester-linked to a peptide (TH19P01) designed to recognize SORT1. Studies in breast and ovarian cancer cell lines have shown that TH1902 exploited SORT1s ligand internalization functions and exerted potent antiproliferative and anti-migratory effects. Within the cell, the docetaxel molecules are released from the conjugate and can then affect polymerization of microtubules leading to aberrant mitosis and apoptosis. Intravenous administration of TH1902 to mice bearing xenografts of MDA-MB-231 breast cancer cells demonstrated a marked superiority of TH1902 over free docetaxel in preventing the growth and relapse of subcutaneous xenografts. In a previous study, SORT1 was reported to have a role in vasculogenic mimicry (VM), and TH1902 was shown to inhibit in vitro VM in these cells and in the ES-2 ovarian cancer cell line.
Click to Show/Hide
|
||||
| Description |
The results show that TH1902 induces more efficiently apoptosis in both cell lines when compared to docetaxel especially in SKOV3 cells. Overall, this suggests that receptor-mediated events appear to account for the increased effects of TH1902 within such a short time frame. To confirm the implication of SORT1 in TH1902 internalization and apoptosis induction, SORT1 was transiently silenced in ES-2 and SKOV3 cells.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 30 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Apotosis rate |
47.00%
|
|||
| Administration Time | 5 h | ||||
| Administration Dosage | 2 μM | ||||
| Evaluation Method | Annexin X/PI staining combined with flow cytometry | ||||
| MOA of PDC |
TH1902 is a peptide-drug conjugate with a payload of two docetaxel molecules ester-linked to a peptide (TH19P01) designed to recognize SORT1. Studies in breast and ovarian cancer cell lines have shown that TH1902 exploited SORT1s ligand internalization functions and exerted potent antiproliferative and anti-migratory effects. Within the cell, the docetaxel molecules are released from the conjugate and can then affect polymerization of microtubules leading to aberrant mitosis and apoptosis. Intravenous administration of TH1902 to mice bearing xenografts of MDA-MB-231 breast cancer cells demonstrated a marked superiority of TH1902 over free docetaxel in preventing the growth and relapse of subcutaneous xenografts. In a previous study, SORT1 was reported to have a role in vasculogenic mimicry (VM), and TH1902 was shown to inhibit in vitro VM in these cells and in the ES-2 ovarian cancer cell line.
Click to Show/Hide
|
||||
| Description |
The results show that TH1902 induces more efficiently apoptosis in both cell lines when compared to docetaxel especially in SKOV3 cells. Overall, this suggests that receptor-mediated events appear to account for the increased effects of TH1902 within such a short time frame. To confirm the implication of SORT1 in TH1902 internalization and apoptosis induction, SORT1 was transiently silenced in ES-2 and SKOV3 cells.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian clear cell adenocarcinoma | ES-2 cell | CVCL_3509 | ||
DTX-P7 [Preclinical]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Non-small cell lung cancer | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
93.2% (Day 28)
|
|||
| Administration Time | Once a week for 4 weeks | ||||
| Administration Dosage | 20 mg/kg | ||||
| Evaluation Method | Tumor volume detection assay | ||||
| MOA of PDC |
Collecitvely, by force of active targeting delivery of DTX via membrane-bound Hsp90, DTX-P7 induces unfolded protein response and subsequent apoptosis by degrading Hsp90, meanwhile awakens and kills the dormant cancer stem cells.
|
||||
| Description |
Intraperitoneal administration of 20 mg/kg DTX-P7 (equivalent to DTX dose calculated as DTX) reduced tumor growth by 93.2% compared with control mice, whereas the tumor growth inhibition of DTX was only 35.9%.
|
||||
| In Vivo Model | A549 cells mouse xenograft model. | ||||
| In Vitro Model | Lung adenocarcinoma | A-549 cell | CVCL_0023 | ||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Non-small cell lung cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
0.62 nM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
Collecitvely, by force of active targeting delivery of DTX via membrane-bound Hsp90, DTX-P7 induces unfolded protein response and subsequent apoptosis by degrading Hsp90, meanwhile awakens and kills the dormant cancer stem cells.
|
||||
| Description |
Intraperitoneal administration of 20 mg/kg DTX-P7 (equivalent to DTX dose calculated as DTX) reduced tumor growth by 93.2% compared with control mice, whereas the tumor growth inhibition of DTX was only 35.9%.
|
||||
| In Vitro Model | Lung adenocarcinoma | H1975 cell | CVCL_1511 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Non-small cell lung cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
11.4 nM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
Collecitvely, by force of active targeting delivery of DTX via membrane-bound Hsp90, DTX-P7 induces unfolded protein response and subsequent apoptosis by degrading Hsp90, meanwhile awakens and kills the dormant cancer stem cells.
|
||||
| Description |
Intraperitoneal administration of 20 mg/kg DTX-P7 (equivalent to DTX dose calculated as DTX) reduced tumor growth by 93.2% compared with control mice, whereas the tumor growth inhibition of DTX was only 35.9%.
|
||||
| In Vitro Model | Lung adenocarcinoma | A-549 cell | CVCL_0023 | ||
BPP-PTX [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
64.00%
|
|||
| Administration Time | 30 days | ||||
| Administration Dosage | 2.4 μmol/kg | ||||
| MOA of PDC |
Here, we report the design, synthesis, and evaluation-in vitro and in vivo-of a novel PDC, namely BPP-PTX, whereby PTX is conjugated to one member of BPPs, Bj-BPP-9a (teprotide), via a succinyl linker. The targeting moiety was carefully selected based on previous studies. It was similarly employed with a nanoparticle carrier in vivo and was shown to modulate improved drug accumulation at the tumor site, thereby curbing tumor growth and extending the lives of tumor-bearing mice. In this study, we demonstrate for the first time that BPP-PTX functions through BPPs cognate receptor, ACE. ACE was overexpressed in TNBC cell lines but not in the receptor-positive cell line. BPP, as part of BPP-PTX, bound ACE and mediated its selective cytotoxic action through this receptor in ACE-positive TNBC cells. Furthermore, BPP-PTX demonstrated improved biodistribution, therapeutic activity, and better safety profile in vivo. These results advocate the significance of BPP-PTX as a suitable tumor-targeting PDC, strongly warranting further refinement and investigations for TNBC.
Click to Show/Hide
|
||||
| Description |
To test the in vivo efficacy and toxicity of BPP-PTX, an orthotopic mouse model was established. ACE-positive MDA-MB-468 cells were injected orthotopically into the mammary fat pad of female nude mice. After the inoculated tumors reached a mean volume of 100 mm3, they were randomly divided into four groups to ensure that the mean tumor volume was evaluated and treated for 28 days with PBS, plain PTX (2.4 μmol/kg, equivalent to 2.0 mg/kg), low-dose BPP-PTX (2.4 μmol/kg, equivalent to 4.9 mg/kg), and high-dose BPP-PTX (9.6 μmol/kg, equivalent to 19.6 mg/kg) every 4 days by i.p. injection. Tumor growth was assessed by measuring tumor volume every 4 days. On the fourth day of testing after the first injection, there was no significant difference in tumor volume compared with control and drug-treated groups. In the subsequent treatments, the tumors of the control mice grew significantly faster than those of mice treated with plain PTX and BPP-PTX. On day 28, mice treated with low-dose BPP-PTX showed an approximately 15% reduction in mean tumor volume compared with mice treated with plain PTX and a 54% reduction compared to controls (PBS). Meanwhile, the mean tumor volumes of mice treated with high-dose BPP-PTX were reduced by 62% compared with control animals and by approximately 30% compared to animals treated with plain PTX. Consistently, the mean tumor weight of mice with low-dose BPP-PTX treatment was 0.20 g (0.17, 0.24), which is lower than that of mice treated with plain PTX [0.26 g (0.21, 0.31)] and control mice [0.43 g (0.37, 0.49)]. The tumor weight of mice treated with high-dose BPP-PTX was significantly lower than that of the control and plain PTX-treated groups, with a mean tumor weight of only 0.16 g (0.14, 0.19). These results suggested that BPP-PTX has good tumor-suppression efficacy in vivo, even better than that of plain PTX.
Click to Show/Hide
|
||||
| In Vivo Model | TNBC nude mouse orthotopic transplantation tumor model. | ||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-468 cell | CVCL_0419 | ||
| Half life period | 5.82 h | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
74.00%
|
|||
| Administration Time | 30 days | ||||
| Administration Dosage | 9.6 μmol/kg | ||||
| MOA of PDC |
Here, we report the design, synthesis, and evaluation-in vitro and in vivo-of a novel PDC, namely BPP-PTX, whereby PTX is conjugated to one member of BPPs, Bj-BPP-9a (teprotide), via a succinyl linker. The targeting moiety was carefully selected based on previous studies. It was similarly employed with a nanoparticle carrier in vivo and was shown to modulate improved drug accumulation at the tumor site, thereby curbing tumor growth and extending the lives of tumor-bearing mice. In this study, we demonstrate for the first time that BPP-PTX functions through BPPs cognate receptor, ACE. ACE was overexpressed in TNBC cell lines but not in the receptor-positive cell line. BPP, as part of BPP-PTX, bound ACE and mediated its selective cytotoxic action through this receptor in ACE-positive TNBC cells. Furthermore, BPP-PTX demonstrated improved biodistribution, therapeutic activity, and better safety profile in vivo. These results advocate the significance of BPP-PTX as a suitable tumor-targeting PDC, strongly warranting further refinement and investigations for TNBC.
Click to Show/Hide
|
||||
| Description |
To test the in vivo efficacy and toxicity of BPP-PTX, an orthotopic mouse model was established. ACE-positive MDA-MB-468 cells were injected orthotopically into the mammary fat pad of female nude mice. After the inoculated tumors reached a mean volume of 100 mm3, they were randomly divided into four groups to ensure that the mean tumor volume was evaluated and treated for 28 days with PBS, plain PTX (2.4 μmol/kg, equivalent to 2.0 mg/kg), low-dose BPP-PTX (2.4 μmol/kg, equivalent to 4.9 mg/kg), and high-dose BPP-PTX (9.6 μmol/kg, equivalent to 19.6 mg/kg) every 4 days by i.p. injection. Tumor growth was assessed by measuring tumor volume every 4 days. On the fourth day of testing after the first injection, there was no significant difference in tumor volume compared with control and drug-treated groups. In the subsequent treatments, the tumors of the control mice grew significantly faster than those of mice treated with plain PTX and BPP-PTX. On day 28, mice treated with low-dose BPP-PTX showed an approximately 15% reduction in mean tumor volume compared with mice treated with plain PTX and a 54% reduction compared to controls (PBS). Meanwhile, the mean tumor volumes of mice treated with high-dose BPP-PTX were reduced by 62% compared with control animals and by approximately 30% compared to animals treated with plain PTX. Consistently, the mean tumor weight of mice with low-dose BPP-PTX treatment was 0.20 g (0.17, 0.24), which is lower than that of mice treated with plain PTX [0.26 g (0.21, 0.31)] and control mice [0.43 g (0.37, 0.49)]. The tumor weight of mice treated with high-dose BPP-PTX was significantly lower than that of the control and plain PTX-treated groups, with a mean tumor weight of only 0.16 g (0.14, 0.19). These results suggested that BPP-PTX has good tumor-suppression efficacy in vivo, even better than that of plain PTX.
Click to Show/Hide
|
||||
| In Vivo Model | TNBC nude mouse orthotopic transplantation tumor model. | ||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-468 cell | CVCL_0419 | ||
| Half life period | 5.82 h | ||||
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Weight loss rate |
1.90%
|
|||
| Administration Dosage | 60 mg/kg | ||||
| MOA of PDC |
Here, we report the design, synthesis, and evaluation-in vitro and in vivo-of a novel PDC, namely BPP-PTX, whereby PTX is conjugated to one member of BPPs, Bj-BPP-9a (teprotide), via a succinyl linker. The targeting moiety was carefully selected based on previous studies. It was similarly employed with a nanoparticle carrier in vivo and was shown to modulate improved drug accumulation at the tumor site, thereby curbing tumor growth and extending the lives of tumor-bearing mice. In this study, we demonstrate for the first time that BPP-PTX functions through BPPs cognate receptor, ACE. ACE was overexpressed in TNBC cell lines but not in the receptor-positive cell line. BPP, as part of BPP-PTX, bound ACE and mediated its selective cytotoxic action through this receptor in ACE-positive TNBC cells. Furthermore, BPP-PTX demonstrated improved biodistribution, therapeutic activity, and better safety profile in vivo. These results advocate the significance of BPP-PTX as a suitable tumor-targeting PDC, strongly warranting further refinement and investigations for TNBC.
Click to Show/Hide
|
||||
| Description |
After obtaining positive in vitro results, BPP-PTX was then tested in vivo. To test the in vivo toxicity of the drug, the maximum tolerated dose (MTD) of BPP-PTX in Balb/c mice was determined. The single-dose MTD (acute toxicity) of plain PTX was 20 mg/kg (equivalent to 23.4 μmol/kg), similar to previous findings in the literature, while the single-dose MTD of BPP-PTX was 100 mg/kg (equivalent to 48.7 μmol/kg). The weight loss was more severe as the drug dose increased. However, the acute weight loss caused by high dose was temporary, with the body weight recovering to baseline levels within 15 days. Then, we evaluated the plasma pharmacokinetics profiles of plain PTX and BPP-PTX. Mice xenografted with MDA-MB-468 received intraperitoneal (i.p.) injections of 15 mg/kg PTX (equivalent to 17.6 μmol/kg) or 36.1 mg/kg BPP-PTX (equivalent to 17.6 μmol/kg). The plasma PTX concentration in the plain PTX-treated group reached peak levels at 1 h and then decreased rapidly, and it was quickly removed from the circulating system. In contrast, the plasma PTX concentration in the BPP-PTX-treated group remained high for an extended period after 1 h, suggesting that BPP-PTX could have a lower release rate in circulation.
Click to Show/Hide
|
||||
| In Vivo Model | Female BALB/c mice. | ||||
| Half life period | 5.82 h | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Weight loss rate |
4.50%
|
|||
| Administration Dosage | 80 mg/kg | ||||
| MOA of PDC |
Here, we report the design, synthesis, and evaluation-in vitro and in vivo-of a novel PDC, namely BPP-PTX, whereby PTX is conjugated to one member of BPPs, Bj-BPP-9a (teprotide), via a succinyl linker. The targeting moiety was carefully selected based on previous studies. It was similarly employed with a nanoparticle carrier in vivo and was shown to modulate improved drug accumulation at the tumor site, thereby curbing tumor growth and extending the lives of tumor-bearing mice. In this study, we demonstrate for the first time that BPP-PTX functions through BPPs cognate receptor, ACE. ACE was overexpressed in TNBC cell lines but not in the receptor-positive cell line. BPP, as part of BPP-PTX, bound ACE and mediated its selective cytotoxic action through this receptor in ACE-positive TNBC cells. Furthermore, BPP-PTX demonstrated improved biodistribution, therapeutic activity, and better safety profile in vivo. These results advocate the significance of BPP-PTX as a suitable tumor-targeting PDC, strongly warranting further refinement and investigations for TNBC.
Click to Show/Hide
|
||||
| Description |
After obtaining positive in vitro results, BPP-PTX was then tested in vivo. To test the in vivo toxicity of the drug, the maximum tolerated dose (MTD) of BPP-PTX in Balb/c mice was determined. The single-dose MTD (acute toxicity) of plain PTX was 20 mg/kg (equivalent to 23.4 μmol/kg), similar to previous findings in the literature, while the single-dose MTD of BPP-PTX was 100 mg/kg (equivalent to 48.7 μmol/kg). The weight loss was more severe as the drug dose increased. However, the acute weight loss caused by high dose was temporary, with the body weight recovering to baseline levels within 15 days. Then, we evaluated the plasma pharmacokinetics profiles of plain PTX and BPP-PTX. Mice xenografted with MDA-MB-468 received intraperitoneal (i.p.) injections of 15 mg/kg PTX (equivalent to 17.6 μmol/kg) or 36.1 mg/kg BPP-PTX (equivalent to 17.6 μmol/kg). The plasma PTX concentration in the plain PTX-treated group reached peak levels at 1 h and then decreased rapidly, and it was quickly removed from the circulating system. In contrast, the plasma PTX concentration in the BPP-PTX-treated group remained high for an extended period after 1 h, suggesting that BPP-PTX could have a lower release rate in circulation.
Click to Show/Hide
|
||||
| In Vivo Model | Female BALB/c mice. | ||||
| Half life period | 5.82 h | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Weight loss rate |
7.20%
|
|||
| Administration Dosage | 100 mg/kg | ||||
| MOA of PDC |
Here, we report the design, synthesis, and evaluation-in vitro and in vivo-of a novel PDC, namely BPP-PTX, whereby PTX is conjugated to one member of BPPs, Bj-BPP-9a (teprotide), via a succinyl linker. The targeting moiety was carefully selected based on previous studies. It was similarly employed with a nanoparticle carrier in vivo and was shown to modulate improved drug accumulation at the tumor site, thereby curbing tumor growth and extending the lives of tumor-bearing mice. In this study, we demonstrate for the first time that BPP-PTX functions through BPPs cognate receptor, ACE. ACE was overexpressed in TNBC cell lines but not in the receptor-positive cell line. BPP, as part of BPP-PTX, bound ACE and mediated its selective cytotoxic action through this receptor in ACE-positive TNBC cells. Furthermore, BPP-PTX demonstrated improved biodistribution, therapeutic activity, and better safety profile in vivo. These results advocate the significance of BPP-PTX as a suitable tumor-targeting PDC, strongly warranting further refinement and investigations for TNBC.
Click to Show/Hide
|
||||
| Description |
After obtaining positive in vitro results, BPP-PTX was then tested in vivo. To test the in vivo toxicity of the drug, the maximum tolerated dose (MTD) of BPP-PTX in Balb/c mice was determined. The single-dose MTD (acute toxicity) of plain PTX was 20 mg/kg (equivalent to 23.4 μmol/kg), similar to previous findings in the literature, while the single-dose MTD of BPP-PTX was 100 mg/kg (equivalent to 48.7 μmol/kg). The weight loss was more severe as the drug dose increased. However, the acute weight loss caused by high dose was temporary, with the body weight recovering to baseline levels within 15 days. Then, we evaluated the plasma pharmacokinetics profiles of plain PTX and BPP-PTX. Mice xenografted with MDA-MB-468 received intraperitoneal (i.p.) injections of 15 mg/kg PTX (equivalent to 17.6 μmol/kg) or 36.1 mg/kg BPP-PTX (equivalent to 17.6 μmol/kg). The plasma PTX concentration in the plain PTX-treated group reached peak levels at 1 h and then decreased rapidly, and it was quickly removed from the circulating system. In contrast, the plasma PTX concentration in the BPP-PTX-treated group remained high for an extended period after 1 h, suggesting that BPP-PTX could have a lower release rate in circulation.
Click to Show/Hide
|
||||
| In Vivo Model | Female BALB/c mice. | ||||
| Half life period | 5.82 h | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Mice survival rate |
75%
|
|||
| Administration Dosage | 120 mg/kg | ||||
| MOA of PDC |
Here, we report the design, synthesis, and evaluation-in vitro and in vivo-of a novel PDC, namely BPP-PTX, whereby PTX is conjugated to one member of BPPs, Bj-BPP-9a (teprotide), via a succinyl linker. The targeting moiety was carefully selected based on previous studies. It was similarly employed with a nanoparticle carrier in vivo and was shown to modulate improved drug accumulation at the tumor site, thereby curbing tumor growth and extending the lives of tumor-bearing mice. In this study, we demonstrate for the first time that BPP-PTX functions through BPPs cognate receptor, ACE. ACE was overexpressed in TNBC cell lines but not in the receptor-positive cell line. BPP, as part of BPP-PTX, bound ACE and mediated its selective cytotoxic action through this receptor in ACE-positive TNBC cells. Furthermore, BPP-PTX demonstrated improved biodistribution, therapeutic activity, and better safety profile in vivo. These results advocate the significance of BPP-PTX as a suitable tumor-targeting PDC, strongly warranting further refinement and investigations for TNBC.
Click to Show/Hide
|
||||
| Description |
After obtaining positive in vitro results, BPP-PTX was then tested in vivo. To test the in vivo toxicity of the drug, the maximum tolerated dose (MTD) of BPP-PTX in Balb/c mice was determined. The single-dose MTD (acute toxicity) of plain PTX was 20 mg/kg (equivalent to 23.4 μmol/kg), similar to previous findings in the literature, while the single-dose MTD of BPP-PTX was 100 mg/kg (equivalent to 48.7 μmol/kg). The weight loss was more severe as the drug dose increased. However, the acute weight loss caused by high dose was temporary, with the body weight recovering to baseline levels within 15 days. Then, we evaluated the plasma pharmacokinetics profiles of plain PTX and BPP-PTX. Mice xenografted with MDA-MB-468 received intraperitoneal (i.p.) injections of 15 mg/kg PTX (equivalent to 17.6 μmol/kg) or 36.1 mg/kg BPP-PTX (equivalent to 17.6 μmol/kg). The plasma PTX concentration in the plain PTX-treated group reached peak levels at 1 h and then decreased rapidly, and it was quickly removed from the circulating system. In contrast, the plasma PTX concentration in the BPP-PTX-treated group remained high for an extended period after 1 h, suggesting that BPP-PTX could have a lower release rate in circulation.
Click to Show/Hide
|
||||
| In Vivo Model | Female BALB/c mice. | ||||
| Half life period | 5.82 h | ||||
| Experiment 5 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Mice survival rate |
100%
|
|||
| Administration Dosage | 60 mg/kg | ||||
| MOA of PDC |
Here, we report the design, synthesis, and evaluation-in vitro and in vivo-of a novel PDC, namely BPP-PTX, whereby PTX is conjugated to one member of BPPs, Bj-BPP-9a (teprotide), via a succinyl linker. The targeting moiety was carefully selected based on previous studies. It was similarly employed with a nanoparticle carrier in vivo and was shown to modulate improved drug accumulation at the tumor site, thereby curbing tumor growth and extending the lives of tumor-bearing mice. In this study, we demonstrate for the first time that BPP-PTX functions through BPPs cognate receptor, ACE. ACE was overexpressed in TNBC cell lines but not in the receptor-positive cell line. BPP, as part of BPP-PTX, bound ACE and mediated its selective cytotoxic action through this receptor in ACE-positive TNBC cells. Furthermore, BPP-PTX demonstrated improved biodistribution, therapeutic activity, and better safety profile in vivo. These results advocate the significance of BPP-PTX as a suitable tumor-targeting PDC, strongly warranting further refinement and investigations for TNBC.
Click to Show/Hide
|
||||
| Description |
After obtaining positive in vitro results, BPP-PTX was then tested in vivo. To test the in vivo toxicity of the drug, the maximum tolerated dose (MTD) of BPP-PTX in Balb/c mice was determined. The single-dose MTD (acute toxicity) of plain PTX was 20 mg/kg (equivalent to 23.4 μmol/kg), similar to previous findings in the literature, while the single-dose MTD of BPP-PTX was 100 mg/kg (equivalent to 48.7 μmol/kg). The weight loss was more severe as the drug dose increased. However, the acute weight loss caused by high dose was temporary, with the body weight recovering to baseline levels within 15 days. Then, we evaluated the plasma pharmacokinetics profiles of plain PTX and BPP-PTX. Mice xenografted with MDA-MB-468 received intraperitoneal (i.p.) injections of 15 mg/kg PTX (equivalent to 17.6 μmol/kg) or 36.1 mg/kg BPP-PTX (equivalent to 17.6 μmol/kg). The plasma PTX concentration in the plain PTX-treated group reached peak levels at 1 h and then decreased rapidly, and it was quickly removed from the circulating system. In contrast, the plasma PTX concentration in the BPP-PTX-treated group remained high for an extended period after 1 h, suggesting that BPP-PTX could have a lower release rate in circulation.
Click to Show/Hide
|
||||
| In Vivo Model | Female BALB/c mice. | ||||
| Half life period | 5.82 h | ||||
| Experiment 6 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Mice survival rate |
100%
|
|||
| Administration Dosage | 80 mg/kg | ||||
| MOA of PDC |
Here, we report the design, synthesis, and evaluation-in vitro and in vivo-of a novel PDC, namely BPP-PTX, whereby PTX is conjugated to one member of BPPs, Bj-BPP-9a (teprotide), via a succinyl linker. The targeting moiety was carefully selected based on previous studies. It was similarly employed with a nanoparticle carrier in vivo and was shown to modulate improved drug accumulation at the tumor site, thereby curbing tumor growth and extending the lives of tumor-bearing mice. In this study, we demonstrate for the first time that BPP-PTX functions through BPPs cognate receptor, ACE. ACE was overexpressed in TNBC cell lines but not in the receptor-positive cell line. BPP, as part of BPP-PTX, bound ACE and mediated its selective cytotoxic action through this receptor in ACE-positive TNBC cells. Furthermore, BPP-PTX demonstrated improved biodistribution, therapeutic activity, and better safety profile in vivo. These results advocate the significance of BPP-PTX as a suitable tumor-targeting PDC, strongly warranting further refinement and investigations for TNBC.
Click to Show/Hide
|
||||
| Description |
After obtaining positive in vitro results, BPP-PTX was then tested in vivo. To test the in vivo toxicity of the drug, the maximum tolerated dose (MTD) of BPP-PTX in Balb/c mice was determined. The single-dose MTD (acute toxicity) of plain PTX was 20 mg/kg (equivalent to 23.4 μmol/kg), similar to previous findings in the literature, while the single-dose MTD of BPP-PTX was 100 mg/kg (equivalent to 48.7 μmol/kg). The weight loss was more severe as the drug dose increased. However, the acute weight loss caused by high dose was temporary, with the body weight recovering to baseline levels within 15 days. Then, we evaluated the plasma pharmacokinetics profiles of plain PTX and BPP-PTX. Mice xenografted with MDA-MB-468 received intraperitoneal (i.p.) injections of 15 mg/kg PTX (equivalent to 17.6 μmol/kg) or 36.1 mg/kg BPP-PTX (equivalent to 17.6 μmol/kg). The plasma PTX concentration in the plain PTX-treated group reached peak levels at 1 h and then decreased rapidly, and it was quickly removed from the circulating system. In contrast, the plasma PTX concentration in the BPP-PTX-treated group remained high for an extended period after 1 h, suggesting that BPP-PTX could have a lower release rate in circulation.
Click to Show/Hide
|
||||
| In Vivo Model | Female BALB/c mice. | ||||
| Half life period | 5.82 h | ||||
| Experiment 7 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Mice survival rate |
100%
|
|||
| Administration Dosage | 100 mg/kg | ||||
| MOA of PDC |
Here, we report the design, synthesis, and evaluation-in vitro and in vivo-of a novel PDC, namely BPP-PTX, whereby PTX is conjugated to one member of BPPs, Bj-BPP-9a (teprotide), via a succinyl linker. The targeting moiety was carefully selected based on previous studies. It was similarly employed with a nanoparticle carrier in vivo and was shown to modulate improved drug accumulation at the tumor site, thereby curbing tumor growth and extending the lives of tumor-bearing mice. In this study, we demonstrate for the first time that BPP-PTX functions through BPPs cognate receptor, ACE. ACE was overexpressed in TNBC cell lines but not in the receptor-positive cell line. BPP, as part of BPP-PTX, bound ACE and mediated its selective cytotoxic action through this receptor in ACE-positive TNBC cells. Furthermore, BPP-PTX demonstrated improved biodistribution, therapeutic activity, and better safety profile in vivo. These results advocate the significance of BPP-PTX as a suitable tumor-targeting PDC, strongly warranting further refinement and investigations for TNBC.
Click to Show/Hide
|
||||
| Description |
After obtaining positive in vitro results, BPP-PTX was then tested in vivo. To test the in vivo toxicity of the drug, the maximum tolerated dose (MTD) of BPP-PTX in Balb/c mice was determined. The single-dose MTD (acute toxicity) of plain PTX was 20 mg/kg (equivalent to 23.4 μmol/kg), similar to previous findings in the literature, while the single-dose MTD of BPP-PTX was 100 mg/kg (equivalent to 48.7 μmol/kg). The weight loss was more severe as the drug dose increased. However, the acute weight loss caused by high dose was temporary, with the body weight recovering to baseline levels within 15 days. Then, we evaluated the plasma pharmacokinetics profiles of plain PTX and BPP-PTX. Mice xenografted with MDA-MB-468 received intraperitoneal (i.p.) injections of 15 mg/kg PTX (equivalent to 17.6 μmol/kg) or 36.1 mg/kg BPP-PTX (equivalent to 17.6 μmol/kg). The plasma PTX concentration in the plain PTX-treated group reached peak levels at 1 h and then decreased rapidly, and it was quickly removed from the circulating system. In contrast, the plasma PTX concentration in the BPP-PTX-treated group remained high for an extended period after 1 h, suggesting that BPP-PTX could have a lower release rate in circulation.
Click to Show/Hide
|
||||
| In Vivo Model | Female BALB/c mice. | ||||
| Half life period | 5.82 h | ||||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
9.5 nM
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Here, we report the design, synthesis, and evaluation-in vitro and in vivo-of a novel PDC, namely BPP-PTX, whereby PTX is conjugated to one member of BPPs, Bj-BPP-9a (teprotide), via a succinyl linker. The targeting moiety was carefully selected based on previous studies. It was similarly employed with a nanoparticle carrier in vivo and was shown to modulate improved drug accumulation at the tumor site, thereby curbing tumor growth and extending the lives of tumor-bearing mice. In this study, we demonstrate for the first time that BPP-PTX functions through BPPs cognate receptor, ACE. ACE was overexpressed in TNBC cell lines but not in the receptor-positive cell line. BPP, as part of BPP-PTX, bound ACE and mediated its selective cytotoxic action through this receptor in ACE-positive TNBC cells. Furthermore, BPP-PTX demonstrated improved biodistribution, therapeutic activity, and better safety profile in vivo. These results advocate the significance of BPP-PTX as a suitable tumor-targeting PDC, strongly warranting further refinement and investigations for TNBC.
Click to Show/Hide
|
||||
| Description |
To evaluate the in vitro antitumor activity, BPP-PTX, uncoupled peptide (BPP), and PTX were under cytotoxicity evaluation by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazole (MTT) assay in ACE-positive TNBC cell lines (MDA-MB-231 and MDA-MB-468) and ACE-negative cell lines (HEK293T). The free BPP peptide did not exhibit any cytotoxicity in all cell lines. In contrast, the IC50 value of BPP-PTX in ACE-negative HEK293T was 616.1 nM [95% CI, (242.7, 1564.0)], which was much higher than that of PTX in the same cell line {6.7 nM [95% CI, (5.3, 8.4)]}. Interestingly, the cytotoxicity of BPP-PTX was comparable with that of PTX in ACE-positive TNBC cell lines. In MDA-MB-231, the IC50 value of BPP-PTX was 9.5 nM [95% CI, (7.0, 12.8)], and that of PTX was 3.1 nM [95% CI, (2.8, 3.5)]. In MDA-MB-468, the IC50 value of BPP-PTX was 12.3 nM [95% CI, (6.8, 22.3)], and that of PTX was 3.0 nM [95% CI, (2.0, 4.7)].
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 (ACE+) cell | CVCL_0062 | ||
| Half life period | 5.82 h | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
12.3 nM
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Here, we report the design, synthesis, and evaluation-in vitro and in vivo-of a novel PDC, namely BPP-PTX, whereby PTX is conjugated to one member of BPPs, Bj-BPP-9a (teprotide), via a succinyl linker. The targeting moiety was carefully selected based on previous studies. It was similarly employed with a nanoparticle carrier in vivo and was shown to modulate improved drug accumulation at the tumor site, thereby curbing tumor growth and extending the lives of tumor-bearing mice. In this study, we demonstrate for the first time that BPP-PTX functions through BPPs cognate receptor, ACE. ACE was overexpressed in TNBC cell lines but not in the receptor-positive cell line. BPP, as part of BPP-PTX, bound ACE and mediated its selective cytotoxic action through this receptor in ACE-positive TNBC cells. Furthermore, BPP-PTX demonstrated improved biodistribution, therapeutic activity, and better safety profile in vivo. These results advocate the significance of BPP-PTX as a suitable tumor-targeting PDC, strongly warranting further refinement and investigations for TNBC.
Click to Show/Hide
|
||||
| Description |
To evaluate the in vitro antitumor activity, BPP-PTX, uncoupled peptide (BPP), and PTX were under cytotoxicity evaluation by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazole (MTT) assay in ACE-positive TNBC cell lines (MDA-MB-231 and MDA-MB-468) and ACE-negative cell lines (HEK293T). The free BPP peptide did not exhibit any cytotoxicity in all cell lines. In contrast, the IC50 value of BPP-PTX in ACE-negative HEK293T was 616.1 nM [95% CI, (242.7, 1564.0)], which was much higher than that of PTX in the same cell line {6.7 nM [95% CI, (5.3, 8.4)]}. Interestingly, the cytotoxicity of BPP-PTX was comparable with that of PTX in ACE-positive TNBC cell lines. In MDA-MB-231, the IC50 value of BPP-PTX was 9.5 nM [95% CI, (7.0, 12.8)], and that of PTX was 3.1 nM [95% CI, (2.8, 3.5)]. In MDA-MB-468, the IC50 value of BPP-PTX was 12.3 nM [95% CI, (6.8, 22.3)], and that of PTX was 3.0 nM [95% CI, (2.0, 4.7)].
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-468 cell | CVCL_0419 | ||
| Half life period | 5.82 h | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
616.1 nM
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Here, we report the design, synthesis, and evaluation-in vitro and in vivo-of a novel PDC, namely BPP-PTX, whereby PTX is conjugated to one member of BPPs, Bj-BPP-9a (teprotide), via a succinyl linker. The targeting moiety was carefully selected based on previous studies. It was similarly employed with a nanoparticle carrier in vivo and was shown to modulate improved drug accumulation at the tumor site, thereby curbing tumor growth and extending the lives of tumor-bearing mice. In this study, we demonstrate for the first time that BPP-PTX functions through BPPs cognate receptor, ACE. ACE was overexpressed in TNBC cell lines but not in the receptor-positive cell line. BPP, as part of BPP-PTX, bound ACE and mediated its selective cytotoxic action through this receptor in ACE-positive TNBC cells. Furthermore, BPP-PTX demonstrated improved biodistribution, therapeutic activity, and better safety profile in vivo. These results advocate the significance of BPP-PTX as a suitable tumor-targeting PDC, strongly warranting further refinement and investigations for TNBC.
Click to Show/Hide
|
||||
| Description |
To evaluate the in vitro antitumor activity, BPP-PTX, uncoupled peptide (BPP), and PTX were under cytotoxicity evaluation by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazole (MTT) assay in ACE-positive TNBC cell lines (MDA-MB-231 and MDA-MB-468) and ACE-negative cell lines (HEK293T). The free BPP peptide did not exhibit any cytotoxicity in all cell lines. In contrast, the IC50 value of BPP-PTX in ACE-negative HEK293T was 616.1 nM [95% CI, (242.7, 1564.0)], which was much higher than that of PTX in the same cell line {6.7 nM [95% CI, (5.3, 8.4)]}. Interestingly, the cytotoxicity of BPP-PTX was comparable with that of PTX in ACE-positive TNBC cell lines. In MDA-MB-231, the IC50 value of BPP-PTX was 9.5 nM [95% CI, (7.0, 12.8)], and that of PTX was 3.1 nM [95% CI, (2.8, 3.5)]. In MDA-MB-468, the IC50 value of BPP-PTX was 12.3 nM [95% CI, (6.8, 22.3)], and that of PTX was 3.0 nM [95% CI, (2.0, 4.7)].
Click to Show/Hide
|
||||
| In Vitro Model | Normal | HEK-293T (ACE-) cell | CVCL_0063 | ||
| Half life period | 5.82 h | ||||
FDPC-NPs [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Tumer volume |
200 mm3
|
|||
| Administration Time | 13 days | ||||
| Administration Dosage | 10 mg DOX/kg | ||||
| MOA of PDC |
In this work, we reported doxorubicin-peptide conjugates (DPCs) with an extracellular tumor acid-responsive sphere-fiber transformation for enhanced residence in tumors. As illustrated in Scheme Scheme1,1, the chemotherapy drug doxorubicin (DOX) was coupled with a peptide (KIGLFRWR) to design a DPC molecule with assembly ability. First, the DPCs, driven by hydrophobic forces from the hydrophobic drug DOX and the IGL fragment, can form spherical DPC nanoparticles (DPC-NPs). Then, along with hydrogen bond between peptides, the aromatic amino acids F and W give the DPC-NPs the ability of self-assembly to DPC-nanofibers (DPC-NFs) due to π-π stacking. The step-by-step assembly process provides opportunities for morphological transformation control. To meet the particle size requirements for intravenous injection, the acid-responsive material 2,3-dimethylmaleic anhydride grafted polylysine, named the functional polylysine graft (FPG), was designed as a shielding layer for DPC-NPs and formed functional doxorubicin-peptide conjugate nanoparticles (FDPC-NPs) by an electrostatic interaction to avoid π-π stacking interactions and hydrogen bond between the DPC-NPs. Therefore, the FDPC-NPs could maintain an appropriate size in blood vessels until entering the tumor stroma by the EPR effect. When the FDPC-NPs passed through the blood vessel and entered the weakly acidic microenvironment of the tumor, the surface potential of the shield was reversed from negative to positive because of acid-sensitive 2,3-dimethylmaleic groups on the FPG. Therefore, FPG would separate from the DPC-NPs because of the mutual repulsion effect from the like charges. Then, DPC-NPs self-assembled into DPC-NFs, thereby staying in the tumor region for a long time. After that, the fibers degraded gradually and free drug penetrated into tumor cells, exerting sustained anti-tumor effect. This study is original and provides new ideas for the design of targeted and long-acting drug delivery systems for tumor therapy.
Click to Show/Hide
|
||||
| Description |
Results showed that DOX solution, DOX-liposomes and FDPC-NPs displayed significant therapeutic effects against tumors (P < 0.001). Particularly, DOX-liposomes and FDPC-NPs behaved better due to the EPR effect.
|
||||
| In Vivo Model | H22 hepatocarcinoma tumor-bearing mouse. | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Percent survival |
95%
|
|||
| Administration Time | 13 days | ||||
| Administration Dosage | 10 mg DOX/kg | ||||
| MOA of PDC |
In this work, we reported doxorubicin-peptide conjugates (DPCs) with an extracellular tumor acid-responsive sphere-fiber transformation for enhanced residence in tumors. As illustrated in Scheme Scheme1,1, the chemotherapy drug doxorubicin (DOX) was coupled with a peptide (KIGLFRWR) to design a DPC molecule with assembly ability. First, the DPCs, driven by hydrophobic forces from the hydrophobic drug DOX and the IGL fragment, can form spherical DPC nanoparticles (DPC-NPs). Then, along with hydrogen bond between peptides, the aromatic amino acids F and W give the DPC-NPs the ability of self-assembly to DPC-nanofibers (DPC-NFs) due to π-π stacking. The step-by-step assembly process provides opportunities for morphological transformation control. To meet the particle size requirements for intravenous injection, the acid-responsive material 2,3-dimethylmaleic anhydride grafted polylysine, named the functional polylysine graft (FPG), was designed as a shielding layer for DPC-NPs and formed functional doxorubicin-peptide conjugate nanoparticles (FDPC-NPs) by an electrostatic interaction to avoid π-π stacking interactions and hydrogen bond between the DPC-NPs. Therefore, the FDPC-NPs could maintain an appropriate size in blood vessels until entering the tumor stroma by the EPR effect. When the FDPC-NPs passed through the blood vessel and entered the weakly acidic microenvironment of the tumor, the surface potential of the shield was reversed from negative to positive because of acid-sensitive 2,3-dimethylmaleic groups on the FPG. Therefore, FPG would separate from the DPC-NPs because of the mutual repulsion effect from the like charges. Then, DPC-NPs self-assembled into DPC-NFs, thereby staying in the tumor region for a long time. After that, the fibers degraded gradually and free drug penetrated into tumor cells, exerting sustained anti-tumor effect. This study is original and provides new ideas for the design of targeted and long-acting drug delivery systems for tumor therapy.
Click to Show/Hide
|
||||
| Description |
Moreover, based on the survivorship curves, treatment with FDPC-NPs remarkably promoted the survival rate of tumor-bearing mice, which furtherly confirmed the therapeutic effect and biological safety of FDPC-NPs.
|
||||
| In Vivo Model | H22 hepatocarcinoma tumor-bearing mouse. | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Body weight |
32g
|
|||
| Administration Time | 13 days | ||||
| Administration Dosage | 10 mg DOX/kg | ||||
| MOA of PDC |
In this work, we reported doxorubicin-peptide conjugates (DPCs) with an extracellular tumor acid-responsive sphere-fiber transformation for enhanced residence in tumors. As illustrated in Scheme Scheme1,1, the chemotherapy drug doxorubicin (DOX) was coupled with a peptide (KIGLFRWR) to design a DPC molecule with assembly ability. First, the DPCs, driven by hydrophobic forces from the hydrophobic drug DOX and the IGL fragment, can form spherical DPC nanoparticles (DPC-NPs). Then, along with hydrogen bond between peptides, the aromatic amino acids F and W give the DPC-NPs the ability of self-assembly to DPC-nanofibers (DPC-NFs) due to π-π stacking. The step-by-step assembly process provides opportunities for morphological transformation control. To meet the particle size requirements for intravenous injection, the acid-responsive material 2,3-dimethylmaleic anhydride grafted polylysine, named the functional polylysine graft (FPG), was designed as a shielding layer for DPC-NPs and formed functional doxorubicin-peptide conjugate nanoparticles (FDPC-NPs) by an electrostatic interaction to avoid π-π stacking interactions and hydrogen bond between the DPC-NPs. Therefore, the FDPC-NPs could maintain an appropriate size in blood vessels until entering the tumor stroma by the EPR effect. When the FDPC-NPs passed through the blood vessel and entered the weakly acidic microenvironment of the tumor, the surface potential of the shield was reversed from negative to positive because of acid-sensitive 2,3-dimethylmaleic groups on the FPG. Therefore, FPG would separate from the DPC-NPs because of the mutual repulsion effect from the like charges. Then, DPC-NPs self-assembled into DPC-NFs, thereby staying in the tumor region for a long time. After that, the fibers degraded gradually and free drug penetrated into tumor cells, exerting sustained anti-tumor effect. This study is original and provides new ideas for the design of targeted and long-acting drug delivery systems for tumor therapy.
Click to Show/Hide
|
||||
| Description |
On the contrary, the administration of DOX-liposomes and FDPC-NPs barely influenced the body weights of the model mice, revealing the safety of DOX-liposomes and FDPC-NPs.
|
||||
| In Vivo Model | H22 hepatocarcinoma tumor-bearing mouse. | ||||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
5.896 μg/mL
|
|||
| MOA of PDC |
In this work, we reported doxorubicin-peptide conjugates (DPCs) with an extracellular tumor acid-responsive sphere-fiber transformation for enhanced residence in tumors. As illustrated in Scheme Scheme1,1, the chemotherapy drug doxorubicin (DOX) was coupled with a peptide (KIGLFRWR) to design a DPC molecule with assembly ability. First, the DPCs, driven by hydrophobic forces from the hydrophobic drug DOX and the IGL fragment, can form spherical DPC nanoparticles (DPC-NPs). Then, along with hydrogen bond between peptides, the aromatic amino acids F and W give the DPC-NPs the ability of self-assembly to DPC-nanofibers (DPC-NFs) due to π-π stacking. The step-by-step assembly process provides opportunities for morphological transformation control. To meet the particle size requirements for intravenous injection, the acid-responsive material 2,3-dimethylmaleic anhydride grafted polylysine, named the functional polylysine graft (FPG), was designed as a shielding layer for DPC-NPs and formed functional doxorubicin-peptide conjugate nanoparticles (FDPC-NPs) by an electrostatic interaction to avoid π-π stacking interactions and hydrogen bond between the DPC-NPs. Therefore, the FDPC-NPs could maintain an appropriate size in blood vessels until entering the tumor stroma by the EPR effect. When the FDPC-NPs passed through the blood vessel and entered the weakly acidic microenvironment of the tumor, the surface potential of the shield was reversed from negative to positive because of acid-sensitive 2,3-dimethylmaleic groups on the FPG. Therefore, FPG would separate from the DPC-NPs because of the mutual repulsion effect from the like charges. Then, DPC-NPs self-assembled into DPC-NFs, thereby staying in the tumor region for a long time. After that, the fibers degraded gradually and free drug penetrated into tumor cells, exerting sustained anti-tumor effect. This study is original and provides new ideas for the design of targeted and long-acting drug delivery systems for tumor therapy.
Click to Show/Hide
|
||||
| Description |
Both the peptide and FPG exhibited no obvious cytotoxicity, while FDPC-NPs and DOX displayed cytotoxicity against tumor cells (IC50 DOX = 2.965 μg/mL; IC50 FDPC-NPs = 5.896 μg/mL) (Figure (Figure6A).6A).
|
||||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Cell viability |
10.00%
|
|||
| Administration Dosage | 50 μg/ml | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In this work, we reported doxorubicin-peptide conjugates (DPCs) with an extracellular tumor acid-responsive sphere-fiber transformation for enhanced residence in tumors. As illustrated in Scheme Scheme1,1, the chemotherapy drug doxorubicin (DOX) was coupled with a peptide (KIGLFRWR) to design a DPC molecule with assembly ability. First, the DPCs, driven by hydrophobic forces from the hydrophobic drug DOX and the IGL fragment, can form spherical DPC nanoparticles (DPC-NPs). Then, along with hydrogen bond between peptides, the aromatic amino acids F and W give the DPC-NPs the ability of self-assembly to DPC-nanofibers (DPC-NFs) due to π-π stacking. The step-by-step assembly process provides opportunities for morphological transformation control. To meet the particle size requirements for intravenous injection, the acid-responsive material 2,3-dimethylmaleic anhydride grafted polylysine, named the functional polylysine graft (FPG), was designed as a shielding layer for DPC-NPs and formed functional doxorubicin-peptide conjugate nanoparticles (FDPC-NPs) by an electrostatic interaction to avoid π-π stacking interactions and hydrogen bond between the DPC-NPs. Therefore, the FDPC-NPs could maintain an appropriate size in blood vessels until entering the tumor stroma by the EPR effect. When the FDPC-NPs passed through the blood vessel and entered the weakly acidic microenvironment of the tumor, the surface potential of the shield was reversed from negative to positive because of acid-sensitive 2,3-dimethylmaleic groups on the FPG. Therefore, FPG would separate from the DPC-NPs because of the mutual repulsion effect from the like charges. Then, DPC-NPs self-assembled into DPC-NFs, thereby staying in the tumor region for a long time. After that, the fibers degraded gradually and free drug penetrated into tumor cells, exerting sustained anti-tumor effect. This study is original and provides new ideas for the design of targeted and long-acting drug delivery systems for tumor therapy.
Click to Show/Hide
|
||||
| Description |
Both the peptide and FPG exhibited no obvious cytotoxicity, while FDPC-NPs and DOX displayed cytotoxicity against tumor cells (IC50 DOX = 2.965 μg/mL; IC50 FDPC-NPs = 5.896 μg/mL) (Figure (Figure6A).6A).
|
||||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Cell viability |
25.00%
|
|||
| Administration Dosage | 25 μg/ml | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In this work, we reported doxorubicin-peptide conjugates (DPCs) with an extracellular tumor acid-responsive sphere-fiber transformation for enhanced residence in tumors. As illustrated in Scheme Scheme1,1, the chemotherapy drug doxorubicin (DOX) was coupled with a peptide (KIGLFRWR) to design a DPC molecule with assembly ability. First, the DPCs, driven by hydrophobic forces from the hydrophobic drug DOX and the IGL fragment, can form spherical DPC nanoparticles (DPC-NPs). Then, along with hydrogen bond between peptides, the aromatic amino acids F and W give the DPC-NPs the ability of self-assembly to DPC-nanofibers (DPC-NFs) due to π-π stacking. The step-by-step assembly process provides opportunities for morphological transformation control. To meet the particle size requirements for intravenous injection, the acid-responsive material 2,3-dimethylmaleic anhydride grafted polylysine, named the functional polylysine graft (FPG), was designed as a shielding layer for DPC-NPs and formed functional doxorubicin-peptide conjugate nanoparticles (FDPC-NPs) by an electrostatic interaction to avoid π-π stacking interactions and hydrogen bond between the DPC-NPs. Therefore, the FDPC-NPs could maintain an appropriate size in blood vessels until entering the tumor stroma by the EPR effect. When the FDPC-NPs passed through the blood vessel and entered the weakly acidic microenvironment of the tumor, the surface potential of the shield was reversed from negative to positive because of acid-sensitive 2,3-dimethylmaleic groups on the FPG. Therefore, FPG would separate from the DPC-NPs because of the mutual repulsion effect from the like charges. Then, DPC-NPs self-assembled into DPC-NFs, thereby staying in the tumor region for a long time. After that, the fibers degraded gradually and free drug penetrated into tumor cells, exerting sustained anti-tumor effect. This study is original and provides new ideas for the design of targeted and long-acting drug delivery systems for tumor therapy.
Click to Show/Hide
|
||||
| Description |
Both the peptide and FPG exhibited no obvious cytotoxicity, while FDPC-NPs and DOX displayed cytotoxicity against tumor cells (IC50 DOX = 2.965 μg/mL; IC50 FDPC-NPs = 5.896 μg/mL) (Figure (Figure6A).6A).
|
||||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Cell viability |
40.00%
|
|||
| Administration Dosage | 10 μg/ml | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In this work, we reported doxorubicin-peptide conjugates (DPCs) with an extracellular tumor acid-responsive sphere-fiber transformation for enhanced residence in tumors. As illustrated in Scheme Scheme1,1, the chemotherapy drug doxorubicin (DOX) was coupled with a peptide (KIGLFRWR) to design a DPC molecule with assembly ability. First, the DPCs, driven by hydrophobic forces from the hydrophobic drug DOX and the IGL fragment, can form spherical DPC nanoparticles (DPC-NPs). Then, along with hydrogen bond between peptides, the aromatic amino acids F and W give the DPC-NPs the ability of self-assembly to DPC-nanofibers (DPC-NFs) due to π-π stacking. The step-by-step assembly process provides opportunities for morphological transformation control. To meet the particle size requirements for intravenous injection, the acid-responsive material 2,3-dimethylmaleic anhydride grafted polylysine, named the functional polylysine graft (FPG), was designed as a shielding layer for DPC-NPs and formed functional doxorubicin-peptide conjugate nanoparticles (FDPC-NPs) by an electrostatic interaction to avoid π-π stacking interactions and hydrogen bond between the DPC-NPs. Therefore, the FDPC-NPs could maintain an appropriate size in blood vessels until entering the tumor stroma by the EPR effect. When the FDPC-NPs passed through the blood vessel and entered the weakly acidic microenvironment of the tumor, the surface potential of the shield was reversed from negative to positive because of acid-sensitive 2,3-dimethylmaleic groups on the FPG. Therefore, FPG would separate from the DPC-NPs because of the mutual repulsion effect from the like charges. Then, DPC-NPs self-assembled into DPC-NFs, thereby staying in the tumor region for a long time. After that, the fibers degraded gradually and free drug penetrated into tumor cells, exerting sustained anti-tumor effect. This study is original and provides new ideas for the design of targeted and long-acting drug delivery systems for tumor therapy.
Click to Show/Hide
|
||||
| Description |
Both the peptide and FPG exhibited no obvious cytotoxicity, while FDPC-NPs and DOX displayed cytotoxicity against tumor cells (IC50 DOX = 2.965 μg/mL; IC50 FDPC-NPs = 5.896 μg/mL) (Figure (Figure6A).6A).
|
||||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Cell viability |
50.00%
|
|||
| Administration Dosage | 5 μg/ml | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In this work, we reported doxorubicin-peptide conjugates (DPCs) with an extracellular tumor acid-responsive sphere-fiber transformation for enhanced residence in tumors. As illustrated in Scheme Scheme1,1, the chemotherapy drug doxorubicin (DOX) was coupled with a peptide (KIGLFRWR) to design a DPC molecule with assembly ability. First, the DPCs, driven by hydrophobic forces from the hydrophobic drug DOX and the IGL fragment, can form spherical DPC nanoparticles (DPC-NPs). Then, along with hydrogen bond between peptides, the aromatic amino acids F and W give the DPC-NPs the ability of self-assembly to DPC-nanofibers (DPC-NFs) due to π-π stacking. The step-by-step assembly process provides opportunities for morphological transformation control. To meet the particle size requirements for intravenous injection, the acid-responsive material 2,3-dimethylmaleic anhydride grafted polylysine, named the functional polylysine graft (FPG), was designed as a shielding layer for DPC-NPs and formed functional doxorubicin-peptide conjugate nanoparticles (FDPC-NPs) by an electrostatic interaction to avoid π-π stacking interactions and hydrogen bond between the DPC-NPs. Therefore, the FDPC-NPs could maintain an appropriate size in blood vessels until entering the tumor stroma by the EPR effect. When the FDPC-NPs passed through the blood vessel and entered the weakly acidic microenvironment of the tumor, the surface potential of the shield was reversed from negative to positive because of acid-sensitive 2,3-dimethylmaleic groups on the FPG. Therefore, FPG would separate from the DPC-NPs because of the mutual repulsion effect from the like charges. Then, DPC-NPs self-assembled into DPC-NFs, thereby staying in the tumor region for a long time. After that, the fibers degraded gradually and free drug penetrated into tumor cells, exerting sustained anti-tumor effect. This study is original and provides new ideas for the design of targeted and long-acting drug delivery systems for tumor therapy.
Click to Show/Hide
|
||||
| Description |
Both the peptide and FPG exhibited no obvious cytotoxicity, while FDPC-NPs and DOX displayed cytotoxicity against tumor cells (IC50 DOX = 2.965 μg/mL; IC50 FDPC-NPs = 5.896 μg/mL) (Figure (Figure6A).6A).
|
||||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Cell viability |
78.00%
|
|||
| Administration Dosage | 2.5 μg/ml | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In this work, we reported doxorubicin-peptide conjugates (DPCs) with an extracellular tumor acid-responsive sphere-fiber transformation for enhanced residence in tumors. As illustrated in Scheme Scheme1,1, the chemotherapy drug doxorubicin (DOX) was coupled with a peptide (KIGLFRWR) to design a DPC molecule with assembly ability. First, the DPCs, driven by hydrophobic forces from the hydrophobic drug DOX and the IGL fragment, can form spherical DPC nanoparticles (DPC-NPs). Then, along with hydrogen bond between peptides, the aromatic amino acids F and W give the DPC-NPs the ability of self-assembly to DPC-nanofibers (DPC-NFs) due to π-π stacking. The step-by-step assembly process provides opportunities for morphological transformation control. To meet the particle size requirements for intravenous injection, the acid-responsive material 2,3-dimethylmaleic anhydride grafted polylysine, named the functional polylysine graft (FPG), was designed as a shielding layer for DPC-NPs and formed functional doxorubicin-peptide conjugate nanoparticles (FDPC-NPs) by an electrostatic interaction to avoid π-π stacking interactions and hydrogen bond between the DPC-NPs. Therefore, the FDPC-NPs could maintain an appropriate size in blood vessels until entering the tumor stroma by the EPR effect. When the FDPC-NPs passed through the blood vessel and entered the weakly acidic microenvironment of the tumor, the surface potential of the shield was reversed from negative to positive because of acid-sensitive 2,3-dimethylmaleic groups on the FPG. Therefore, FPG would separate from the DPC-NPs because of the mutual repulsion effect from the like charges. Then, DPC-NPs self-assembled into DPC-NFs, thereby staying in the tumor region for a long time. After that, the fibers degraded gradually and free drug penetrated into tumor cells, exerting sustained anti-tumor effect. This study is original and provides new ideas for the design of targeted and long-acting drug delivery systems for tumor therapy.
Click to Show/Hide
|
||||
| Description |
Both the peptide and FPG exhibited no obvious cytotoxicity, while FDPC-NPs and DOX displayed cytotoxicity against tumor cells (IC50 DOX = 2.965 μg/mL; IC50 FDPC-NPs = 5.896 μg/mL) (Figure (Figure6A).6A).
|
||||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Cell viability |
85.00%
|
|||
| Administration Dosage | 1 μg/ml | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In this work, we reported doxorubicin-peptide conjugates (DPCs) with an extracellular tumor acid-responsive sphere-fiber transformation for enhanced residence in tumors. As illustrated in Scheme Scheme1,1, the chemotherapy drug doxorubicin (DOX) was coupled with a peptide (KIGLFRWR) to design a DPC molecule with assembly ability. First, the DPCs, driven by hydrophobic forces from the hydrophobic drug DOX and the IGL fragment, can form spherical DPC nanoparticles (DPC-NPs). Then, along with hydrogen bond between peptides, the aromatic amino acids F and W give the DPC-NPs the ability of self-assembly to DPC-nanofibers (DPC-NFs) due to π-π stacking. The step-by-step assembly process provides opportunities for morphological transformation control. To meet the particle size requirements for intravenous injection, the acid-responsive material 2,3-dimethylmaleic anhydride grafted polylysine, named the functional polylysine graft (FPG), was designed as a shielding layer for DPC-NPs and formed functional doxorubicin-peptide conjugate nanoparticles (FDPC-NPs) by an electrostatic interaction to avoid π-π stacking interactions and hydrogen bond between the DPC-NPs. Therefore, the FDPC-NPs could maintain an appropriate size in blood vessels until entering the tumor stroma by the EPR effect. When the FDPC-NPs passed through the blood vessel and entered the weakly acidic microenvironment of the tumor, the surface potential of the shield was reversed from negative to positive because of acid-sensitive 2,3-dimethylmaleic groups on the FPG. Therefore, FPG would separate from the DPC-NPs because of the mutual repulsion effect from the like charges. Then, DPC-NPs self-assembled into DPC-NFs, thereby staying in the tumor region for a long time. After that, the fibers degraded gradually and free drug penetrated into tumor cells, exerting sustained anti-tumor effect. This study is original and provides new ideas for the design of targeted and long-acting drug delivery systems for tumor therapy.
Click to Show/Hide
|
||||
| Description |
Both the peptide and FPG exhibited no obvious cytotoxicity, while FDPC-NPs and DOX displayed cytotoxicity against tumor cells (IC50 DOX = 2.965 μg/mL; IC50 FDPC-NPs = 5.896 μg/mL) (Figure (Figure6A).6A).
|
||||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Cell viability |
98.00%
|
|||
| Administration Dosage | 0.1 μg/ml | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In this work, we reported doxorubicin-peptide conjugates (DPCs) with an extracellular tumor acid-responsive sphere-fiber transformation for enhanced residence in tumors. As illustrated in Scheme Scheme1,1, the chemotherapy drug doxorubicin (DOX) was coupled with a peptide (KIGLFRWR) to design a DPC molecule with assembly ability. First, the DPCs, driven by hydrophobic forces from the hydrophobic drug DOX and the IGL fragment, can form spherical DPC nanoparticles (DPC-NPs). Then, along with hydrogen bond between peptides, the aromatic amino acids F and W give the DPC-NPs the ability of self-assembly to DPC-nanofibers (DPC-NFs) due to π-π stacking. The step-by-step assembly process provides opportunities for morphological transformation control. To meet the particle size requirements for intravenous injection, the acid-responsive material 2,3-dimethylmaleic anhydride grafted polylysine, named the functional polylysine graft (FPG), was designed as a shielding layer for DPC-NPs and formed functional doxorubicin-peptide conjugate nanoparticles (FDPC-NPs) by an electrostatic interaction to avoid π-π stacking interactions and hydrogen bond between the DPC-NPs. Therefore, the FDPC-NPs could maintain an appropriate size in blood vessels until entering the tumor stroma by the EPR effect. When the FDPC-NPs passed through the blood vessel and entered the weakly acidic microenvironment of the tumor, the surface potential of the shield was reversed from negative to positive because of acid-sensitive 2,3-dimethylmaleic groups on the FPG. Therefore, FPG would separate from the DPC-NPs because of the mutual repulsion effect from the like charges. Then, DPC-NPs self-assembled into DPC-NFs, thereby staying in the tumor region for a long time. After that, the fibers degraded gradually and free drug penetrated into tumor cells, exerting sustained anti-tumor effect. This study is original and provides new ideas for the design of targeted and long-acting drug delivery systems for tumor therapy.
Click to Show/Hide
|
||||
| Description |
Both the peptide and FPG exhibited no obvious cytotoxicity, while FDPC-NPs and DOX displayed cytotoxicity against tumor cells (IC50 DOX = 2.965 μg/mL; IC50 FDPC-NPs = 5.896 μg/mL) (Figure (Figure6A).6A).
|
||||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Apotosis rate |
22.00%
|
|||
| Administration Dosage | 2 μM | ||||
| MOA of PDC |
In this work, we reported doxorubicin-peptide conjugates (DPCs) with an extracellular tumor acid-responsive sphere-fiber transformation for enhanced residence in tumors. As illustrated in Scheme Scheme1,1, the chemotherapy drug doxorubicin (DOX) was coupled with a peptide (KIGLFRWR) to design a DPC molecule with assembly ability. First, the DPCs, driven by hydrophobic forces from the hydrophobic drug DOX and the IGL fragment, can form spherical DPC nanoparticles (DPC-NPs). Then, along with hydrogen bond between peptides, the aromatic amino acids F and W give the DPC-NPs the ability of self-assembly to DPC-nanofibers (DPC-NFs) due to π-π stacking. The step-by-step assembly process provides opportunities for morphological transformation control. To meet the particle size requirements for intravenous injection, the acid-responsive material 2,3-dimethylmaleic anhydride grafted polylysine, named the functional polylysine graft (FPG), was designed as a shielding layer for DPC-NPs and formed functional doxorubicin-peptide conjugate nanoparticles (FDPC-NPs) by an electrostatic interaction to avoid π-π stacking interactions and hydrogen bond between the DPC-NPs. Therefore, the FDPC-NPs could maintain an appropriate size in blood vessels until entering the tumor stroma by the EPR effect. When the FDPC-NPs passed through the blood vessel and entered the weakly acidic microenvironment of the tumor, the surface potential of the shield was reversed from negative to positive because of acid-sensitive 2,3-dimethylmaleic groups on the FPG. Therefore, FPG would separate from the DPC-NPs because of the mutual repulsion effect from the like charges. Then, DPC-NPs self-assembled into DPC-NFs, thereby staying in the tumor region for a long time. After that, the fibers degraded gradually and free drug penetrated into tumor cells, exerting sustained anti-tumor effect. This study is original and provides new ideas for the design of targeted and long-acting drug delivery systems for tumor therapy.
Click to Show/Hide
|
||||
| Description |
Additionally, the results from flow cytometry with annexin-V-FITC/PI double staining showed that FDPC-NPs and DOX significantly increased the proportion of apoptotic cells in a concentration-dependent manner, and the two groups exhibited similar cytotoxicity.
|
||||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Apotosis rate |
30.00%
|
|||
| Administration Dosage | 10 μM | ||||
| MOA of PDC |
In this work, we reported doxorubicin-peptide conjugates (DPCs) with an extracellular tumor acid-responsive sphere-fiber transformation for enhanced residence in tumors. As illustrated in Scheme Scheme1,1, the chemotherapy drug doxorubicin (DOX) was coupled with a peptide (KIGLFRWR) to design a DPC molecule with assembly ability. First, the DPCs, driven by hydrophobic forces from the hydrophobic drug DOX and the IGL fragment, can form spherical DPC nanoparticles (DPC-NPs). Then, along with hydrogen bond between peptides, the aromatic amino acids F and W give the DPC-NPs the ability of self-assembly to DPC-nanofibers (DPC-NFs) due to π-π stacking. The step-by-step assembly process provides opportunities for morphological transformation control. To meet the particle size requirements for intravenous injection, the acid-responsive material 2,3-dimethylmaleic anhydride grafted polylysine, named the functional polylysine graft (FPG), was designed as a shielding layer for DPC-NPs and formed functional doxorubicin-peptide conjugate nanoparticles (FDPC-NPs) by an electrostatic interaction to avoid π-π stacking interactions and hydrogen bond between the DPC-NPs. Therefore, the FDPC-NPs could maintain an appropriate size in blood vessels until entering the tumor stroma by the EPR effect. When the FDPC-NPs passed through the blood vessel and entered the weakly acidic microenvironment of the tumor, the surface potential of the shield was reversed from negative to positive because of acid-sensitive 2,3-dimethylmaleic groups on the FPG. Therefore, FPG would separate from the DPC-NPs because of the mutual repulsion effect from the like charges. Then, DPC-NPs self-assembled into DPC-NFs, thereby staying in the tumor region for a long time. After that, the fibers degraded gradually and free drug penetrated into tumor cells, exerting sustained anti-tumor effect. This study is original and provides new ideas for the design of targeted and long-acting drug delivery systems for tumor therapy.
Click to Show/Hide
|
||||
| Description |
Additionally, the results from flow cytometry with annexin-V-FITC/PI double staining showed that FDPC-NPs and DOX significantly increased the proportion of apoptotic cells in a concentration-dependent manner, and the two groups exhibited similar cytotoxicity.
|
||||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Apotosis rate |
40.00%
|
|||
| Administration Dosage | 20 μM | ||||
| MOA of PDC |
In this work, we reported doxorubicin-peptide conjugates (DPCs) with an extracellular tumor acid-responsive sphere-fiber transformation for enhanced residence in tumors. As illustrated in Scheme Scheme1,1, the chemotherapy drug doxorubicin (DOX) was coupled with a peptide (KIGLFRWR) to design a DPC molecule with assembly ability. First, the DPCs, driven by hydrophobic forces from the hydrophobic drug DOX and the IGL fragment, can form spherical DPC nanoparticles (DPC-NPs). Then, along with hydrogen bond between peptides, the aromatic amino acids F and W give the DPC-NPs the ability of self-assembly to DPC-nanofibers (DPC-NFs) due to π-π stacking. The step-by-step assembly process provides opportunities for morphological transformation control. To meet the particle size requirements for intravenous injection, the acid-responsive material 2,3-dimethylmaleic anhydride grafted polylysine, named the functional polylysine graft (FPG), was designed as a shielding layer for DPC-NPs and formed functional doxorubicin-peptide conjugate nanoparticles (FDPC-NPs) by an electrostatic interaction to avoid π-π stacking interactions and hydrogen bond between the DPC-NPs. Therefore, the FDPC-NPs could maintain an appropriate size in blood vessels until entering the tumor stroma by the EPR effect. When the FDPC-NPs passed through the blood vessel and entered the weakly acidic microenvironment of the tumor, the surface potential of the shield was reversed from negative to positive because of acid-sensitive 2,3-dimethylmaleic groups on the FPG. Therefore, FPG would separate from the DPC-NPs because of the mutual repulsion effect from the like charges. Then, DPC-NPs self-assembled into DPC-NFs, thereby staying in the tumor region for a long time. After that, the fibers degraded gradually and free drug penetrated into tumor cells, exerting sustained anti-tumor effect. This study is original and provides new ideas for the design of targeted and long-acting drug delivery systems for tumor therapy.
Click to Show/Hide
|
||||
| Description |
Additionally, the results from flow cytometry with annexin-V-FITC/PI double staining showed that FDPC-NPs and DOX significantly increased the proportion of apoptotic cells in a concentration-dependent manner, and the two groups exhibited similar cytotoxicity.
|
||||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
M1-PTX [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [9] | ||||
| Indication | Glioma | ||||
| Efficacy Data | Percent survival |
0%
|
|||
| Administration Time | 25 days | ||||
| Administration Dosage | Normalized to 17.3 mg/kg PTX | ||||
| Description |
The survival of the PTX group was markedly lower than that of the Vehicle group. M1-PTX did not improve survival, whereas the survival of the M1-RGD-PTX group was increased.
|
||||
| In Vivo Model | U87MG-Luc-bearing xenograft model. | ||||
| In Vitro Model | Glioblastoma | U87MG-Luc cell | CVCL_5J15 | ||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [9] | ||||
| Indication | Glioma | ||||
| Efficacy Data | Cell viability |
60%
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 100 nM | ||||
| Evaluation Method | MTS assay | ||||
| Description |
Both PDCs showed higher in vitro cytotoxicity than free PTX.
|
||||
| In Vitro Model | Glioblastoma | U-87MG cell | CVCL_0022 | ||
M1-RGD-PTX [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [9] | ||||
| Indication | Glioma | ||||
| Efficacy Data | Percent survival |
20%
|
|||
| Administration Time | 25 days | ||||
| Administration Dosage | Normalized to 17.3 mg/kg PTX | ||||
| Description |
The survival of the PTX group was markedly lower than that of the Vehicle group. M1-PTX did not improve survival, whereas the survival of the M1-RGD-PTX group was increased.
|
||||
| In Vivo Model | U87MG-Luc-bearing xenograft model. | ||||
| In Vitro Model | Glioblastoma | U87MG-Luc cell | CVCL_5J15 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [9] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Percent survival |
50%
|
|||
| Administration Time | 20 days | ||||
| Administration Dosage | 44 mg/kg | ||||
| Description |
PTX did not increase survival, while the PDC M1-RGD-PTX markedly increased survival.
|
||||
| In Vivo Model | MDA-MB-231BR mouse model. | ||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [9] | ||||
| Indication | Glioma | ||||
| Efficacy Data | Median lethal dose (LD50) |
152 mg/kg
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 100 nM | ||||
| Description |
The fitted LD50 for M1-RGD-PTX was 152 mg/kg (equivalent to 60 mg/kg PTX).
|
||||
| In Vitro Model | Glioblastoma | U-87MG cell | CVCL_0022 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [9] | ||||
| Indication | Glioma | ||||
| Efficacy Data | Cell viability |
55%
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 100 nM | ||||
| Evaluation Method | MTS assay | ||||
| Description |
Both PDCs showed higher in vitro cytotoxicity than free PTX.
|
||||
| In Vitro Model | Glioblastoma | U-87MG cell | CVCL_0022 | ||
DOXKGFRWR [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [10] | ||||
| Indication | Hepatocellular carcinoma | ||||
| Efficacy Data | Tumor volume |
376 mm3
|
|||
| Description |
The antitumor efficacy of the DOX-KGFRWR nanofiber was superior to all other treatments, with a final tumor volume of 376 mm3.
|
||||
| In Vivo Model | SMMC7721 tumorbearing mice. | ||||
| Half life period | 24.52±13.17 h | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [10] | ||||
| Indication | Hepatocellular carcinoma | ||||
| Efficacy Data | Percent survival |
50%
|
|||
| Administration Time | 40 days | ||||
| Description |
DOX-KGFRWR has significantly prolonged survival rates.
|
||||
| In Vivo Model | SMMC7721 tumorbearing mice. | ||||
| Half life period | 24.52±13.17 h | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [10] | ||||
| Indication | Hepatocellular carcinoma | ||||
| Efficacy Data | Percent survival | > 40 days | |||
| Administration Time | 30 days | ||||
| Description |
DOX-KGFRWR has significantly prolonged survival rates.
|
||||
| In Vivo Model | SMMC7721 tumorbearing mice. | ||||
| Half life period | 24.52±13.17 h | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [10] | ||||
| Indication | Hepatocellular carcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
5.27 µM
|
|||
| Description |
The IC50 values for KGFRWR, DOX, and DOX-KGFRWR against the MMP2 enzyme were 14.19, 8.68, and 5.27 10-6 m, respectively.
|
||||
| In Vivo Model | SMMC7721 pulmonary metastatic mouse model. | ||||
| Half life period | 24.52±13.17 h | ||||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [10] | ||||
| Indication | Hepatocellular carcinoma | ||||
| Efficacy Data | Migration rates |
19.90%
|
|||
| Description |
Cells treated with DOX and DOX-KGFRWR exhibited markedly decreased migration, with migration rates of 39.4% and 19.9%, respectively, compared with those in the control, indicating that DOX-KGWRFR exerted a stronger inhibiting effect on migration than DOX.
|
||||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
| Half life period | 24.52±13.17 h | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [10] | ||||
| Indication | Hepatocellular carcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
34.55 µg mL-1
|
|||
| Evaluation Method | MTT assay | ||||
| Description |
The IC50 values for DOX-KGFRWR is 34.55 μg mL-1.
|
||||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
| Half life period | 24.52±13.17 h | ||||
NLG-RGD [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [11] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Percentage of Treg cells |
1%
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 25 µM | ||||
| Evaluation Method | Quantification of tumor-infiltrating immune cells assay | ||||
| MOA of PDC |
Significantly, this nanoinhibitor boosts the antitumor immune response of PD-L1 blockade by increasing immune effector cells and reducing immunosuppressive cells.
|
||||
| Description |
These findings support that aPD-L5 plus NLG-RGD NI efficiently elicits antitumor immunity by enhancing the activation and survival of immune effector cells and attenuating the accumulation immunosuppressive Treg cells.
|
||||
| In Vivo Model | Pan02 tumor model. | ||||
| In Vitro Model | Normal | Regulatory CD4+ T cell | Homo sapiens | ||
| Experiment 2 Reporting the Activity Data of This PDC | [11] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Percentage of NK cells |
2%
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 25 µM | ||||
| Evaluation Method | Quantification of tumor-infiltrating immune cells assay | ||||
| MOA of PDC |
Significantly, this nanoinhibitor boosts the antitumor immune response of PD-L1 blockade by increasing immune effector cells and reducing immunosuppressive cells.
|
||||
| Description |
These findings support that aPD-L6 plus NLG-RGD NI efficiently elicits antitumor immunity by enhancing the activation and survival of immune effector cells and attenuating the accumulation immunosuppressive Treg cells.
|
||||
| In Vivo Model | Pan02 tumor model. | ||||
| In Vitro Model | Normal | Natural killer cell | Homo sapiens | ||
| Experiment 3 Reporting the Activity Data of This PDC | [11] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Percentage of IFN-γ-producing CD8 T cells |
40%
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 25 µM | ||||
| Evaluation Method | Quantification of tumor-infiltrating immune cells assay | ||||
| MOA of PDC |
Significantly, this nanoinhibitor boosts the antitumor immune response of PD-L1 blockade by increasing immune effector cells and reducing immunosuppressive cells.
|
||||
| Description |
These findings support that aPD-L4 plus NLG-RGD NI efficiently elicits antitumor immunity by enhancing the activation and survival of immune effector cells and attenuating the accumulation immunosuppressive Treg cells.
|
||||
| In Vivo Model | Pan02 tumor model. | ||||
| In Vitro Model | Normal | IFN-gamma-producing CD4 T cell | Homo sapiens | ||
| Experiment 4 Reporting the Activity Data of This PDC | [11] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Percentage of IFN-γ-producing CD4 T cells |
16%
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 25 µM | ||||
| Evaluation Method | Quantification of tumor-infiltrating immune cells assay | ||||
| MOA of PDC |
Significantly, this nanoinhibitor boosts the antitumor immune response of PD-L1 blockade by increasing immune effector cells and reducing immunosuppressive cells.
|
||||
| Description |
These findings support that aPD-L3 plus NLG-RGD NI efficiently elicits antitumor immunity by enhancing the activation and survival of immune effector cells and attenuating the accumulation immunosuppressive Treg cells.
|
||||
| In Vivo Model | Pan02 tumor model. | ||||
| In Vitro Model | Normal | IFN-gamma-producing CD4 T cell | Homo sapiens | ||
| Experiment 5 Reporting the Activity Data of This PDC | [11] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Percentage of CD8 T cells |
34%
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 25 µM | ||||
| Evaluation Method | Quantification of tumor-infiltrating immune cells assay | ||||
| MOA of PDC |
Significantly, this nanoinhibitor boosts the antitumor immune response of PD-L1 blockade by increasing immune effector cells and reducing immunosuppressive cells.
|
||||
| Description |
These findings support that aPD-L2 plus NLG-RGD NI efficiently elicits antitumor immunity by enhancing the activation and survival of immune effector cells and attenuating the accumulation immunosuppressive Treg cells.
|
||||
| In Vivo Model | Pan02 tumor model. | ||||
| In Vitro Model | Normal | CD8 T cell | Homo sapiens | ||
| Experiment 6 Reporting the Activity Data of This PDC | [11] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Percentage of CD4 T cells |
32%
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 25 µM | ||||
| Evaluation Method | Quantification of tumor-infiltrating immune cells assay | ||||
| MOA of PDC |
Significantly, this nanoinhibitor boosts the antitumor immune response of PD-L1 blockade by increasing immune effector cells and reducing immunosuppressive cells.
|
||||
| Description |
These findings support that aPD-L1 plus NLG-RGD NI efficiently elicits antitumor immunity by enhancing the activation and survival of immune effector cells and attenuating the accumulation immunosuppressive Treg cells.
|
||||
| In Vivo Model | Pan02 tumor model. | ||||
| In Vitro Model | Normal | CD4 T cell | Homo sapiens | ||
Cq-C4-TP10 [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [12] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
0.8 ± 0.1 µM
|
|||
| MOA of PDC |
The significant increase in the hemolytic activity of TP10 upon conjugation to the 4-aminoquinoline suggests that drug cargo prevents an otherwise active CPP carrier from exerting the desired cell penetrating/antiplasmodial action safely, as it produces conjugates that exert membranolytic activity.
|
||||
| In Vivo Model | Plasmodium falciparum 3D7. | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [12] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
1.5 µM
|
|||
| MOA of PDC |
The significant increase in the hemolytic activity of TP10 upon conjugation to the 4-aminoquinoline suggests that drug cargo prevents an otherwise active CPP carrier from exerting the desired cell penetrating/antiplasmodial action safely, as it produces conjugates that exert membranolytic activity.
|
||||
| Description |
Only three of the Cq-C4-CPP conjugates, namely, 5a, 5b, and 5g, displayed IC50 values below 10 μM, with TP10- and Transportan-derived conjugates 5a (IC50 = 1.52 μM) and 5b (IC50 = 5.20 μM) being the most active.
|
||||
| In Vivo Model | Plasmodium falciparum W2. | ||||
Cq-C4-Transportan [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [12] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
5.2 µM
|
|||
| MOA of PDC |
The significant increase in the hemolytic activity of TP10 upon conjugation to the 4-aminoquinoline suggests that drug cargo prevents an otherwise active CPP carrier from exerting the desired cell penetrating/antiplasmodial action safely, as it produces conjugates that exert membranolytic activity.
|
||||
| Description |
Only three of the Cq-C4-CPP conjugates, namely, 5a, 5b, and 5g, displayed IC50 values below 10 μM, with TP10- and Transportan-derived conjugates 5a (IC50 = 1.52 μM) and 5b (IC50 = 5.20 μM) being the most active.
|
||||
| In Vivo Model | Plasmodium falciparum W2. | ||||
Cq-C4-PasTAT [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [12] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
8.5 µM
|
|||
| MOA of PDC |
The significant increase in the hemolytic activity of TP10 upon conjugation to the 4-aminoquinoline suggests that drug cargo prevents an otherwise active CPP carrier from exerting the desired cell penetrating/antiplasmodial action safely, as it produces conjugates that exert membranolytic activity.
|
||||
| Description |
Only three of the Cq-C4-CPP conjugates, namely, 5a, 5b, and 5g, displayed IC50 values below 10 μM, with TP10- and Transportan-derived conjugates 5a (IC50 = 1.52 μM) and 5b (IC50 = 5.20 μM) being the most active.
|
||||
| In Vivo Model | Plasmodium falciparum W2. | ||||
Cq-C4-DPT-sh1 [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [12] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 10 µM | |||
| MOA of PDC |
The significant increase in the hemolytic activity of TP10 upon conjugation to the 4-aminoquinoline suggests that drug cargo prevents an otherwise active CPP carrier from exerting the desired cell penetrating/antiplasmodial action safely, as it produces conjugates that exert membranolytic activity.
|
||||
| Description |
Only three of the Cq-C4-CPP conjugates, namely, 5a, 5b, and 5g, displayed IC50 values below 10 μM, with TP10- and Transportan-derived conjugates 5a (IC50 = 1.52 μM) and 5b (IC50 = 5.20 μM) being the most active.
|
||||
| In Vivo Model | Plasmodium falciparum W2. | ||||
Cq-C4-DPT-sh2 [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [12] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 10 µM | |||
| MOA of PDC |
The significant increase in the hemolytic activity of TP10 upon conjugation to the 4-aminoquinoline suggests that drug cargo prevents an otherwise active CPP carrier from exerting the desired cell penetrating/antiplasmodial action safely, as it produces conjugates that exert membranolytic activity.
|
||||
| Description |
Only three of the Cq-C4-CPP conjugates, namely, 5a, 5b, and 5g, displayed IC50 values below 10 μM, with TP10- and Transportan-derived conjugates 5a (IC50 = 1.52 μM) and 5b (IC50 = 5.20 μM) being the most active.
|
||||
| In Vivo Model | Plasmodium falciparum W2. | ||||
Cq-C4-IDR-1018 [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [12] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 10 µM | |||
| MOA of PDC |
The significant increase in the hemolytic activity of TP10 upon conjugation to the 4-aminoquinoline suggests that drug cargo prevents an otherwise active CPP carrier from exerting the desired cell penetrating/antiplasmodial action safely, as it produces conjugates that exert membranolytic activity.
|
||||
| Description |
Only three of the Cq-C4-CPP conjugates, namely, 5a, 5b, and 5g, displayed IC50 values below 10 μM, with TP10- and Transportan-derived conjugates 5a (IC50 = 1.52 μM) and 5b (IC50 = 5.20 μM) being the most active.
|
||||
| In Vivo Model | Plasmodium falciparum W2. | ||||
Cq-C4-TAT [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [12] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 10 µM | |||
| MOA of PDC |
The significant increase in the hemolytic activity of TP10 upon conjugation to the 4-aminoquinoline suggests that drug cargo prevents an otherwise active CPP carrier from exerting the desired cell penetrating/antiplasmodial action safely, as it produces conjugates that exert membranolytic activity.
|
||||
| Description |
Only three of the Cq-C4-CPP conjugates, namely, 5a, 5b, and 5g, displayed IC50 values below 10 μM, with TP10- and Transportan-derived conjugates 5a (IC50 = 1.52 μM) and 5b (IC50 = 5.20 μM) being the most active.
|
||||
| In Vivo Model | Plasmodium falciparum W2. | ||||
Cq-C4-R9 [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [12] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 10 µM | |||
| MOA of PDC |
The significant increase in the hemolytic activity of TP10 upon conjugation to the 4-aminoquinoline suggests that drug cargo prevents an otherwise active CPP carrier from exerting the desired cell penetrating/antiplasmodial action safely, as it produces conjugates that exert membranolytic activity.
|
||||
| Description |
Only three of the Cq-C4-CPP conjugates, namely, 5a, 5b, and 5g, displayed IC50 values below 10 μM, with TP10- and Transportan-derived conjugates 5a (IC50 = 1.52 μM) and 5b (IC50 = 5.20 μM) being the most active.
|
||||
| In Vivo Model | Plasmodium falciparum W2. | ||||
Cq-C4-Penetratin [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [12] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 10 µM | |||
| MOA of PDC |
The significant increase in the hemolytic activity of TP10 upon conjugation to the 4-aminoquinoline suggests that drug cargo prevents an otherwise active CPP carrier from exerting the desired cell penetrating/antiplasmodial action safely, as it produces conjugates that exert membranolytic activity.
|
||||
| Description |
Only three of the Cq-C4-CPP conjugates, namely, 5a, 5b, and 5g, displayed IC50 values below 10 μM, with TP10- and Transportan-derived conjugates 5a (IC50 = 1.52 μM) and 5b (IC50 = 5.20 μM) being the most active.
|
||||
| In Vivo Model | Plasmodium falciparum W2. | ||||
Antimicrobial peptides (AMP) 1 - Tobramycin conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Selectivity index |
20.66
|
|||
| MOA of PDC |
We have synthesized a series of MAAPCs incorporating tobramycin with variations in the composition, sequence, and conjugation site of the peptide transporter. The MAAPCs exhibit good selectivity for bacterial cell membranes over mammalian cell membranes and do not induce any significant hemolysis of human red blood cells. MAAPCs exhibit better antibacterial activity against actively growing Gram-negative E. coli (MIC < 15 μM) than actively growing Gram-positive S. aureus (MIC < 25 μM). MAAPC01 exhibits the highest permeabilization of the outer membrane, with all other MAAPCs showing less permeation activity; tobramycin exhibits no outer membrane activity. MAAPC01 and MAAPC05 exhibit the highest inner membrane permeability, comparable to the control melittin. MAAPC02 and 03 exhibit less membrane activity, and MAAPC04 and tobramycin show negligible inner membrane activity. Higher levels of membrane activity correlate well with antimicrobial activity against persisters, where MAAPC01 and MAAPC05 show much better activity than tobramycin alone or MAAPC04.
Click to Show/Hide
|
||||
| Description |
One of the limitations to the clinical use of AMPs as antimicrobial agents is their hemolytic activity which is associated with increased toxicity. Thus, as a first assessment of the MAAPC toxicity, we investigated whether the conjugates may cause lysis of human red blood cells (hRBCs). Notably, all MAAPCs exhibited negligible hemolytic activity against hRBCs (HC50 > 500 μM), which is the first indicator of the MAAPC safety toward eukaryotic cells. Bacterial and animal cell membranes significantly differ in composition. Indeed, microbial cell surfaces contain more anionic lipids (overall negatively charged) whereas mammalian cell membranes have more lipids with neutral zwitterionic head groups (overall neutrally charged). The MAAPCs were designed to selectively discriminate between bacterial and mammalian cells. The selectivity indexes (SIs), defined as HC50/MIC, for E. coli and S. aureus demonstrated great selectivity of all MAAPCs to bacteria over hRBCs, which implies that the MAAPCs are effective against bacteria without causing harm to human cells. MAAPC04 displayed the highest bacterial selectivity (SI > 640 and SI > 160 against E. coli and S. aureus, respectively) likely due to its reduced hydrophobic content.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Selectivity index |
82.64
|
|||
| MOA of PDC |
We have synthesized a series of MAAPCs incorporating tobramycin with variations in the composition, sequence, and conjugation site of the peptide transporter. The MAAPCs exhibit good selectivity for bacterial cell membranes over mammalian cell membranes and do not induce any significant hemolysis of human red blood cells. MAAPCs exhibit better antibacterial activity against actively growing Gram-negative E. coli (MIC < 15 μM) than actively growing Gram-positive S. aureus (MIC < 25 μM). MAAPC01 exhibits the highest permeabilization of the outer membrane, with all other MAAPCs showing less permeation activity; tobramycin exhibits no outer membrane activity. MAAPC01 and MAAPC05 exhibit the highest inner membrane permeability, comparable to the control melittin. MAAPC02 and 03 exhibit less membrane activity, and MAAPC04 and tobramycin show negligible inner membrane activity. Higher levels of membrane activity correlate well with antimicrobial activity against persisters, where MAAPC01 and MAAPC05 show much better activity than tobramycin alone or MAAPC04.
Click to Show/Hide
|
||||
| Description |
One of the limitations to the clinical use of AMPs as antimicrobial agents is their hemolytic activity which is associated with increased toxicity. Thus, as a first assessment of the MAAPC toxicity, we investigated whether the conjugates may cause lysis of human red blood cells (hRBCs). Notably, all MAAPCs exhibited negligible hemolytic activity against hRBCs (HC50 > 500 μM), which is the first indicator of the MAAPC safety toward eukaryotic cells. Bacterial and animal cell membranes significantly differ in composition. Indeed, microbial cell surfaces contain more anionic lipids (overall negatively charged) whereas mammalian cell membranes have more lipids with neutral zwitterionic head groups (overall neutrally charged). The MAAPCs were designed to selectively discriminate between bacterial and mammalian cells. The selectivity indexes (SIs), defined as HC50/MIC, for E. coli and S. aureus demonstrated great selectivity of all MAAPCs to bacteria over hRBCs, which implies that the MAAPCs are effective against bacteria without causing harm to human cells. MAAPC04 displayed the highest bacterial selectivity (SI > 640 and SI > 160 against E. coli and S. aureus, respectively) likely due to its reduced hydrophobic content.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 511145 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
6.3 ± 0.0 μM
|
|||
| Administration Time | 18 h | ||||
| MOA of PDC |
We have synthesized a series of MAAPCs incorporating tobramycin with variations in the composition, sequence, and conjugation site of the peptide transporter. The MAAPCs exhibit good selectivity for bacterial cell membranes over mammalian cell membranes and do not induce any significant hemolysis of human red blood cells. MAAPCs exhibit better antibacterial activity against actively growing Gram-negative E. coli (MIC < 15 μM) than actively growing Gram-positive S. aureus (MIC < 25 μM). MAAPC01 exhibits the highest permeabilization of the outer membrane, with all other MAAPCs showing less permeation activity; tobramycin exhibits no outer membrane activity. MAAPC01 and MAAPC05 exhibit the highest inner membrane permeability, comparable to the control melittin. MAAPC02 and 03 exhibit less membrane activity, and MAAPC04 and tobramycin show negligible inner membrane activity. Higher levels of membrane activity correlate well with antimicrobial activity against persisters, where MAAPC01 and MAAPC05 show much better activity than tobramycin alone or MAAPC04.
Click to Show/Hide
|
||||
| Description |
We first investigated the antimicrobial potency of these new MAAPCs by determining their minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC) against Escherichia coli (E. coli MG1655) and Staphylococcus aureus (SA113) pathogens with a turbidity-based microdilution broth assay. Overall, all the MAAPCs display good antimicrobial activity and tend to be more selective to E. coli (MIC range 3.1-12.5 μM) over S. aureus (MIC range 12.5-25 μM), which follows the general observation that tobramycin is particularly active against Gram-negative organisms. Their MBC values are either equal to or slightly higher than their respective MIC values suggesting that the compounds not only are inhibiting bacterial growth but are also bactericidal. In contrast, the unconjugated peptides (P1-P4) exhibited no or little antimicrobial activity against E. coli and S. aureus. However, the MAAPCs did not show better activity than tobramycin, which is expected because our conjugates are not designed to target lab strains but instead to have high efficacy against clinically relevant pathogens such as persisters, resistant bacteria, and anaerobes that are impermeable to existing antibiotics. It should be noted that a mixture of the peptide itself with tobramycin does not enhance the antimicrobial activity compared to tobramycin alone, indicating there is no synergistic effect between the two entities when simply mixed. Whereas MAAPC05 showed similar activity against S. aureus in comparison to MAAPC01 (indicating that the terminus of conjugation did not greatly impact activity), it displayed a 2-fold increase of the MIC against E. coli. In contrast, MAAPC02, MAAPC03, and MAAPC04 displayed improved activity compared to MAAPC01. These results indicate the importance of the peptide transporter sequence as well as the conjugation site for tobramycin. Interestingly, MAAPC02 and MAAPC03 have nearly identical amino-acid composition as MAAPC01 (the only difference is a single glycine), but the amino acids are in a different sequence. In the MAAPC02 analog, the hydrophobic amino acids are clustered along one side of the -helical wheel of the peptide, which confers higher amphiphilic character and greater helical propensity. In the MAAPC03 analog, the peptide has the reversed sequence of MAAPC01. There are examples in the literature that the reversed analogs of antimicrobial peptides possess equal or enhanced antimicrobial activities. The amphiphilicity and peptide sequence orientation might play a role on the membrane-peptide interaction. However, further experiments are required to elucidate the reasons for this improved antibacterial activity.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 511145 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
25.0 ± 0.0 μM
|
|||
| Administration Time | 18 h | ||||
| MOA of PDC |
We have synthesized a series of MAAPCs incorporating tobramycin with variations in the composition, sequence, and conjugation site of the peptide transporter. The MAAPCs exhibit good selectivity for bacterial cell membranes over mammalian cell membranes and do not induce any significant hemolysis of human red blood cells. MAAPCs exhibit better antibacterial activity against actively growing Gram-negative E. coli (MIC < 15 μM) than actively growing Gram-positive S. aureus (MIC < 25 μM). MAAPC01 exhibits the highest permeabilization of the outer membrane, with all other MAAPCs showing less permeation activity; tobramycin exhibits no outer membrane activity. MAAPC01 and MAAPC05 exhibit the highest inner membrane permeability, comparable to the control melittin. MAAPC02 and 03 exhibit less membrane activity, and MAAPC04 and tobramycin show negligible inner membrane activity. Higher levels of membrane activity correlate well with antimicrobial activity against persisters, where MAAPC01 and MAAPC05 show much better activity than tobramycin alone or MAAPC04.
Click to Show/Hide
|
||||
| Description |
We first investigated the antimicrobial potency of these new MAAPCs by determining their minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC) against Escherichia coli (E. coli MG1655) and Staphylococcus aureus (SA113) pathogens with a turbidity-based microdilution broth assay. Overall, all the MAAPCs display good antimicrobial activity and tend to be more selective to E. coli (MIC range 3.1-12.5 μM) over S. aureus (MIC range 12.5-25 μM), which follows the general observation that tobramycin is particularly active against Gram-negative organisms. Their MBC values are either equal to or slightly higher than their respective MIC values suggesting that the compounds not only are inhibiting bacterial growth but are also bactericidal. In contrast, the unconjugated peptides (P1-P4) exhibited no or little antimicrobial activity against E. coli and S. aureus. However, the MAAPCs did not show better activity than tobramycin, which is expected because our conjugates are not designed to target lab strains but instead to have high efficacy against clinically relevant pathogens such as persisters, resistant bacteria, and anaerobes that are impermeable to existing antibiotics. It should be noted that a mixture of the peptide itself with tobramycin does not enhance the antimicrobial activity compared to tobramycin alone, indicating there is no synergistic effect between the two entities when simply mixed. Whereas MAAPC05 showed similar activity against S. aureus in comparison to MAAPC01 (indicating that the terminus of conjugation did not greatly impact activity), it displayed a 2-fold increase of the MIC against E. coli. In contrast, MAAPC02, MAAPC03, and MAAPC04 displayed improved activity compared to MAAPC01. These results indicate the importance of the peptide transporter sequence as well as the conjugation site for tobramycin. Interestingly, MAAPC02 and MAAPC03 have nearly identical amino-acid composition as MAAPC01 (the only difference is a single glycine), but the amino acids are in a different sequence. In the MAAPC02 analog, the hydrophobic amino acids are clustered along one side of the -helical wheel of the peptide, which confers higher amphiphilic character and greater helical propensity. In the MAAPC03 analog, the peptide has the reversed sequence of MAAPC01. There are examples in the literature that the reversed analogs of antimicrobial peptides possess equal or enhanced antimicrobial activities. The amphiphilicity and peptide sequence orientation might play a role on the membrane-peptide interaction. However, further experiments are required to elucidate the reasons for this improved antibacterial activity.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum bactericidal concentration (MBC) |
6.3 ± 0.0 μM
|
|||
| Administration Time | 18 h | ||||
| MOA of PDC |
We have synthesized a series of MAAPCs incorporating tobramycin with variations in the composition, sequence, and conjugation site of the peptide transporter. The MAAPCs exhibit good selectivity for bacterial cell membranes over mammalian cell membranes and do not induce any significant hemolysis of human red blood cells. MAAPCs exhibit better antibacterial activity against actively growing Gram-negative E. coli (MIC < 15 μM) than actively growing Gram-positive S. aureus (MIC < 25 μM). MAAPC01 exhibits the highest permeabilization of the outer membrane, with all other MAAPCs showing less permeation activity; tobramycin exhibits no outer membrane activity. MAAPC01 and MAAPC05 exhibit the highest inner membrane permeability, comparable to the control melittin. MAAPC02 and 03 exhibit less membrane activity, and MAAPC04 and tobramycin show negligible inner membrane activity. Higher levels of membrane activity correlate well with antimicrobial activity against persisters, where MAAPC01 and MAAPC05 show much better activity than tobramycin alone or MAAPC04.
Click to Show/Hide
|
||||
| Description |
We first investigated the antimicrobial potency of these new MAAPCs by determining their minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC) against Escherichia coli (E. coli MG1655) and Staphylococcus aureus (SA113) pathogens with a turbidity-based microdilution broth assay. Overall, all the MAAPCs display good antimicrobial activity and tend to be more selective to E. coli (MIC range 3.1-12.5 μM) over S. aureus (MIC range 12.5-25 μM), which follows the general observation that tobramycin is particularly active against Gram-negative organisms. Their MBC values are either equal to or slightly higher than their respective MIC values suggesting that the compounds not only are inhibiting bacterial growth but are also bactericidal. In contrast, the unconjugated peptides (P1-P4) exhibited no or little antimicrobial activity against E. coli and S. aureus. However, the MAAPCs did not show better activity than tobramycin, which is expected because our conjugates are not designed to target lab strains but instead to have high efficacy against clinically relevant pathogens such as persisters, resistant bacteria, and anaerobes that are impermeable to existing antibiotics. It should be noted that a mixture of the peptide itself with tobramycin does not enhance the antimicrobial activity compared to tobramycin alone, indicating there is no synergistic effect between the two entities when simply mixed. Whereas MAAPC05 showed similar activity against S. aureus in comparison to MAAPC01 (indicating that the terminus of conjugation did not greatly impact activity), it displayed a 2-fold increase of the MIC against E. coli. In contrast, MAAPC02, MAAPC03, and MAAPC04 displayed improved activity compared to MAAPC01. These results indicate the importance of the peptide transporter sequence as well as the conjugation site for tobramycin. Interestingly, MAAPC02 and MAAPC03 have nearly identical amino-acid composition as MAAPC01 (the only difference is a single glycine), but the amino acids are in a different sequence. In the MAAPC02 analog, the hydrophobic amino acids are clustered along one side of the -helical wheel of the peptide, which confers higher amphiphilic character and greater helical propensity. In the MAAPC03 analog, the peptide has the reversed sequence of MAAPC01. There are examples in the literature that the reversed analogs of antimicrobial peptides possess equal or enhanced antimicrobial activities. The amphiphilicity and peptide sequence orientation might play a role on the membrane-peptide interaction. However, further experiments are required to elucidate the reasons for this improved antibacterial activity.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 511145 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum bactericidal concentration (MBC) |
33.3 ± 14.4 μM
|
|||
| Administration Time | 18 h | ||||
| MOA of PDC |
We have synthesized a series of MAAPCs incorporating tobramycin with variations in the composition, sequence, and conjugation site of the peptide transporter. The MAAPCs exhibit good selectivity for bacterial cell membranes over mammalian cell membranes and do not induce any significant hemolysis of human red blood cells. MAAPCs exhibit better antibacterial activity against actively growing Gram-negative E. coli (MIC < 15 μM) than actively growing Gram-positive S. aureus (MIC < 25 μM). MAAPC01 exhibits the highest permeabilization of the outer membrane, with all other MAAPCs showing less permeation activity; tobramycin exhibits no outer membrane activity. MAAPC01 and MAAPC05 exhibit the highest inner membrane permeability, comparable to the control melittin. MAAPC02 and 03 exhibit less membrane activity, and MAAPC04 and tobramycin show negligible inner membrane activity. Higher levels of membrane activity correlate well with antimicrobial activity against persisters, where MAAPC01 and MAAPC05 show much better activity than tobramycin alone or MAAPC04.
Click to Show/Hide
|
||||
| Description |
We first investigated the antimicrobial potency of these new MAAPCs by determining their minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC) against Escherichia coli (E. coli MG1655) and Staphylococcus aureus (SA113) pathogens with a turbidity-based microdilution broth assay. Overall, all the MAAPCs display good antimicrobial activity and tend to be more selective to E. coli (MIC range 3.1-12.5 μM) over S. aureus (MIC range 12.5-25 μM), which follows the general observation that tobramycin is particularly active against Gram-negative organisms. Their MBC values are either equal to or slightly higher than their respective MIC values suggesting that the compounds not only are inhibiting bacterial growth but are also bactericidal. In contrast, the unconjugated peptides (P1-P4) exhibited no or little antimicrobial activity against E. coli and S. aureus. However, the MAAPCs did not show better activity than tobramycin, which is expected because our conjugates are not designed to target lab strains but instead to have high efficacy against clinically relevant pathogens such as persisters, resistant bacteria, and anaerobes that are impermeable to existing antibiotics. It should be noted that a mixture of the peptide itself with tobramycin does not enhance the antimicrobial activity compared to tobramycin alone, indicating there is no synergistic effect between the two entities when simply mixed. Whereas MAAPC05 showed similar activity against S. aureus in comparison to MAAPC01 (indicating that the terminus of conjugation did not greatly impact activity), it displayed a 2-fold increase of the MIC against E. coli. In contrast, MAAPC02, MAAPC03, and MAAPC04 displayed improved activity compared to MAAPC01. These results indicate the importance of the peptide transporter sequence as well as the conjugation site for tobramycin. Interestingly, MAAPC02 and MAAPC03 have nearly identical amino-acid composition as MAAPC01 (the only difference is a single glycine), but the amino acids are in a different sequence. In the MAAPC02 analog, the hydrophobic amino acids are clustered along one side of the -helical wheel of the peptide, which confers higher amphiphilic character and greater helical propensity. In the MAAPC03 analog, the peptide has the reversed sequence of MAAPC01. There are examples in the literature that the reversed analogs of antimicrobial peptides possess equal or enhanced antimicrobial activities. The amphiphilicity and peptide sequence orientation might play a role on the membrane-peptide interaction. However, further experiments are required to elucidate the reasons for this improved antibacterial activity.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
516.5 μM
|
|||
| Administration Time | 1 h | ||||
| MOA of PDC |
We have synthesized a series of MAAPCs incorporating tobramycin with variations in the composition, sequence, and conjugation site of the peptide transporter. The MAAPCs exhibit good selectivity for bacterial cell membranes over mammalian cell membranes and do not induce any significant hemolysis of human red blood cells. MAAPCs exhibit better antibacterial activity against actively growing Gram-negative E. coli (MIC < 15 μM) than actively growing Gram-positive S. aureus (MIC < 25 μM). MAAPC01 exhibits the highest permeabilization of the outer membrane, with all other MAAPCs showing less permeation activity; tobramycin exhibits no outer membrane activity. MAAPC01 and MAAPC05 exhibit the highest inner membrane permeability, comparable to the control melittin. MAAPC02 and 03 exhibit less membrane activity, and MAAPC04 and tobramycin show negligible inner membrane activity. Higher levels of membrane activity correlate well with antimicrobial activity against persisters, where MAAPC01 and MAAPC05 show much better activity than tobramycin alone or MAAPC04.
Click to Show/Hide
|
||||
| Description |
One of the limitations to the clinical use of AMPs as antimicrobial agents is their hemolytic activity which is associated with increased toxicity. Thus, as a first assessment of the MAAPC toxicity, we investigated whether the conjugates may cause lysis of human red blood cells (hRBCs). Notably, all MAAPCs exhibited negligible hemolytic activity against hRBCs (HC50 > 500 μM), which is the first indicator of the MAAPC safety toward eukaryotic cells. Bacterial and animal cell membranes significantly differ in composition. Indeed, microbial cell surfaces contain more anionic lipids (overall negatively charged) whereas mammalian cell membranes have more lipids with neutral zwitterionic head groups (overall neutrally charged). The MAAPCs were designed to selectively discriminate between bacterial and mammalian cells. The selectivity indexes (SIs), defined as HC50/MIC, for E. coli and S. aureus demonstrated great selectivity of all MAAPCs to bacteria over hRBCs, which implies that the MAAPCs are effective against bacteria without causing harm to human cells. MAAPC04 displayed the highest bacterial selectivity (SI > 640 and SI > 160 against E. coli and S. aureus, respectively) likely due to its reduced hydrophobic content.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Human red blood cells | Homo sapiens | ||
Antimicrobial peptides (AMP) 4 - Tobramycin conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Selectivity index | > 80 | |||
| MOA of PDC |
We have synthesized a series of MAAPCs incorporating tobramycin with variations in the composition, sequence, and conjugation site of the peptide transporter. The MAAPCs exhibit good selectivity for bacterial cell membranes over mammalian cell membranes and do not induce any significant hemolysis of human red blood cells. MAAPCs exhibit better antibacterial activity against actively growing Gram-negative E. coli (MIC < 15 μM) than actively growing Gram-positive S. aureus (MIC < 25 μM). MAAPC01 exhibits the highest permeabilization of the outer membrane, with all other MAAPCs showing less permeation activity; tobramycin exhibits no outer membrane activity. MAAPC01 and MAAPC05 exhibit the highest inner membrane permeability, comparable to the control melittin. MAAPC02 and 03 exhibit less membrane activity, and MAAPC04 and tobramycin show negligible inner membrane activity. Higher levels of membrane activity correlate well with antimicrobial activity against persisters, where MAAPC01 and MAAPC05 show much better activity than tobramycin alone or MAAPC04.
Click to Show/Hide
|
||||
| Description |
One of the limitations to the clinical use of AMPs as antimicrobial agents is their hemolytic activity which is associated with increased toxicity. Thus, as a first assessment of the MAAPC toxicity, we investigated whether the conjugates may cause lysis of human red blood cells (hRBCs). Notably, all MAAPCs exhibited negligible hemolytic activity against hRBCs (HC50 > 500 μM), which is the first indicator of the MAAPC safety toward eukaryotic cells. Bacterial and animal cell membranes significantly differ in composition. Indeed, microbial cell surfaces contain more anionic lipids (overall negatively charged) whereas mammalian cell membranes have more lipids with neutral zwitterionic head groups (overall neutrally charged). The MAAPCs were designed to selectively discriminate between bacterial and mammalian cells. The selectivity indexes (SIs), defined as HC50/MIC, for E. coli and S. aureus demonstrated great selectivity of all MAAPCs to bacteria over hRBCs, which implies that the MAAPCs are effective against bacteria without causing harm to human cells. MAAPC04 displayed the highest bacterial selectivity (SI > 640 and SI > 160 against E. coli and S. aureus, respectively) likely due to its reduced hydrophobic content.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Selectivity index | > 320 | |||
| MOA of PDC |
We have synthesized a series of MAAPCs incorporating tobramycin with variations in the composition, sequence, and conjugation site of the peptide transporter. The MAAPCs exhibit good selectivity for bacterial cell membranes over mammalian cell membranes and do not induce any significant hemolysis of human red blood cells. MAAPCs exhibit better antibacterial activity against actively growing Gram-negative E. coli (MIC < 15 μM) than actively growing Gram-positive S. aureus (MIC < 25 μM). MAAPC01 exhibits the highest permeabilization of the outer membrane, with all other MAAPCs showing less permeation activity; tobramycin exhibits no outer membrane activity. MAAPC01 and MAAPC05 exhibit the highest inner membrane permeability, comparable to the control melittin. MAAPC02 and 03 exhibit less membrane activity, and MAAPC04 and tobramycin show negligible inner membrane activity. Higher levels of membrane activity correlate well with antimicrobial activity against persisters, where MAAPC01 and MAAPC05 show much better activity than tobramycin alone or MAAPC04.
Click to Show/Hide
|
||||
| Description |
One of the limitations to the clinical use of AMPs as antimicrobial agents is their hemolytic activity which is associated with increased toxicity. Thus, as a first assessment of the MAAPC toxicity, we investigated whether the conjugates may cause lysis of human red blood cells (hRBCs). Notably, all MAAPCs exhibited negligible hemolytic activity against hRBCs (HC50 > 500 μM), which is the first indicator of the MAAPC safety toward eukaryotic cells. Bacterial and animal cell membranes significantly differ in composition. Indeed, microbial cell surfaces contain more anionic lipids (overall negatively charged) whereas mammalian cell membranes have more lipids with neutral zwitterionic head groups (overall neutrally charged). The MAAPCs were designed to selectively discriminate between bacterial and mammalian cells. The selectivity indexes (SIs), defined as HC50/MIC, for E. coli and S. aureus demonstrated great selectivity of all MAAPCs to bacteria over hRBCs, which implies that the MAAPCs are effective against bacteria without causing harm to human cells. MAAPC04 displayed the highest bacterial selectivity (SI > 640 and SI > 160 against E. coli and S. aureus, respectively) likely due to its reduced hydrophobic content.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 511145 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
3.1 ± 0.0 μM
|
|||
| Administration Time | 18 h | ||||
| MOA of PDC |
We have synthesized a series of MAAPCs incorporating tobramycin with variations in the composition, sequence, and conjugation site of the peptide transporter. The MAAPCs exhibit good selectivity for bacterial cell membranes over mammalian cell membranes and do not induce any significant hemolysis of human red blood cells. MAAPCs exhibit better antibacterial activity against actively growing Gram-negative E. coli (MIC < 15 μM) than actively growing Gram-positive S. aureus (MIC < 25 μM). MAAPC01 exhibits the highest permeabilization of the outer membrane, with all other MAAPCs showing less permeation activity; tobramycin exhibits no outer membrane activity. MAAPC01 and MAAPC05 exhibit the highest inner membrane permeability, comparable to the control melittin. MAAPC02 and 03 exhibit less membrane activity, and MAAPC04 and tobramycin show negligible inner membrane activity. Higher levels of membrane activity correlate well with antimicrobial activity against persisters, where MAAPC01 and MAAPC05 show much better activity than tobramycin alone or MAAPC04.
Click to Show/Hide
|
||||
| Description |
We first investigated the antimicrobial potency of these new MAAPCs by determining their minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC) against Escherichia coli (E. coli MG1655) and Staphylococcus aureus (SA113) pathogens with a turbidity-based microdilution broth assay. Overall, all the MAAPCs display good antimicrobial activity and tend to be more selective to E. coli (MIC range 3.1-12.5 μM) over S. aureus (MIC range 12.5-25 μM), which follows the general observation that tobramycin is particularly active against Gram-negative organisms. Their MBC values are either equal to or slightly higher than their respective MIC values suggesting that the compounds not only are inhibiting bacterial growth but are also bactericidal. In contrast, the unconjugated peptides (P1-P4) exhibited no or little antimicrobial activity against E. coli and S. aureus. However, the MAAPCs did not show better activity than tobramycin, which is expected because our conjugates are not designed to target lab strains but instead to have high efficacy against clinically relevant pathogens such as persisters, resistant bacteria, and anaerobes that are impermeable to existing antibiotics. It should be noted that a mixture of the peptide itself with tobramycin does not enhance the antimicrobial activity compared to tobramycin alone, indicating there is no synergistic effect between the two entities when simply mixed. Whereas MAAPC05 showed similar activity against S. aureus in comparison to MAAPC01 (indicating that the terminus of conjugation did not greatly impact activity), it displayed a 2-fold increase of the MIC against E. coli. In contrast, MAAPC02, MAAPC03, and MAAPC04 displayed improved activity compared to MAAPC01. These results indicate the importance of the peptide transporter sequence as well as the conjugation site for tobramycin. Interestingly, MAAPC02 and MAAPC03 have nearly identical amino-acid composition as MAAPC01 (the only difference is a single glycine), but the amino acids are in a different sequence. In the MAAPC02 analog, the hydrophobic amino acids are clustered along one side of the -helical wheel of the peptide, which confers higher amphiphilic character and greater helical propensity. In the MAAPC03 analog, the peptide has the reversed sequence of MAAPC01. There are examples in the literature that the reversed analogs of antimicrobial peptides possess equal or enhanced antimicrobial activities. The amphiphilicity and peptide sequence orientation might play a role on the membrane-peptide interaction. However, further experiments are required to elucidate the reasons for this improved antibacterial activity.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 511145 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
12.5 ± 0.0 μM
|
|||
| Administration Time | 18 h | ||||
| MOA of PDC |
We have synthesized a series of MAAPCs incorporating tobramycin with variations in the composition, sequence, and conjugation site of the peptide transporter. The MAAPCs exhibit good selectivity for bacterial cell membranes over mammalian cell membranes and do not induce any significant hemolysis of human red blood cells. MAAPCs exhibit better antibacterial activity against actively growing Gram-negative E. coli (MIC < 15 μM) than actively growing Gram-positive S. aureus (MIC < 25 μM). MAAPC01 exhibits the highest permeabilization of the outer membrane, with all other MAAPCs showing less permeation activity; tobramycin exhibits no outer membrane activity. MAAPC01 and MAAPC05 exhibit the highest inner membrane permeability, comparable to the control melittin. MAAPC02 and 03 exhibit less membrane activity, and MAAPC04 and tobramycin show negligible inner membrane activity. Higher levels of membrane activity correlate well with antimicrobial activity against persisters, where MAAPC01 and MAAPC05 show much better activity than tobramycin alone or MAAPC04.
Click to Show/Hide
|
||||
| Description |
We first investigated the antimicrobial potency of these new MAAPCs by determining their minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC) against Escherichia coli (E. coli MG1655) and Staphylococcus aureus (SA113) pathogens with a turbidity-based microdilution broth assay. Overall, all the MAAPCs display good antimicrobial activity and tend to be more selective to E. coli (MIC range 3.1-12.5 μM) over S. aureus (MIC range 12.5-25 μM), which follows the general observation that tobramycin is particularly active against Gram-negative organisms. Their MBC values are either equal to or slightly higher than their respective MIC values suggesting that the compounds not only are inhibiting bacterial growth but are also bactericidal. In contrast, the unconjugated peptides (P1-P4) exhibited no or little antimicrobial activity against E. coli and S. aureus. However, the MAAPCs did not show better activity than tobramycin, which is expected because our conjugates are not designed to target lab strains but instead to have high efficacy against clinically relevant pathogens such as persisters, resistant bacteria, and anaerobes that are impermeable to existing antibiotics. It should be noted that a mixture of the peptide itself with tobramycin does not enhance the antimicrobial activity compared to tobramycin alone, indicating there is no synergistic effect between the two entities when simply mixed. Whereas MAAPC05 showed similar activity against S. aureus in comparison to MAAPC01 (indicating that the terminus of conjugation did not greatly impact activity), it displayed a 2-fold increase of the MIC against E. coli. In contrast, MAAPC02, MAAPC03, and MAAPC04 displayed improved activity compared to MAAPC01. These results indicate the importance of the peptide transporter sequence as well as the conjugation site for tobramycin. Interestingly, MAAPC02 and MAAPC03 have nearly identical amino-acid composition as MAAPC01 (the only difference is a single glycine), but the amino acids are in a different sequence. In the MAAPC02 analog, the hydrophobic amino acids are clustered along one side of the -helical wheel of the peptide, which confers higher amphiphilic character and greater helical propensity. In the MAAPC03 analog, the peptide has the reversed sequence of MAAPC01. There are examples in the literature that the reversed analogs of antimicrobial peptides possess equal or enhanced antimicrobial activities. The amphiphilicity and peptide sequence orientation might play a role on the membrane-peptide interaction. However, further experiments are required to elucidate the reasons for this improved antibacterial activity.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum bactericidal concentration (MBC) |
3.1 ± 0.0 μM
|
|||
| Administration Time | 18 h | ||||
| MOA of PDC |
We have synthesized a series of MAAPCs incorporating tobramycin with variations in the composition, sequence, and conjugation site of the peptide transporter. The MAAPCs exhibit good selectivity for bacterial cell membranes over mammalian cell membranes and do not induce any significant hemolysis of human red blood cells. MAAPCs exhibit better antibacterial activity against actively growing Gram-negative E. coli (MIC < 15 μM) than actively growing Gram-positive S. aureus (MIC < 25 μM). MAAPC01 exhibits the highest permeabilization of the outer membrane, with all other MAAPCs showing less permeation activity; tobramycin exhibits no outer membrane activity. MAAPC01 and MAAPC05 exhibit the highest inner membrane permeability, comparable to the control melittin. MAAPC02 and 03 exhibit less membrane activity, and MAAPC04 and tobramycin show negligible inner membrane activity. Higher levels of membrane activity correlate well with antimicrobial activity against persisters, where MAAPC01 and MAAPC05 show much better activity than tobramycin alone or MAAPC04.
Click to Show/Hide
|
||||
| Description |
We first investigated the antimicrobial potency of these new MAAPCs by determining their minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC) against Escherichia coli (E. coli MG1655) and Staphylococcus aureus (SA113) pathogens with a turbidity-based microdilution broth assay. Overall, all the MAAPCs display good antimicrobial activity and tend to be more selective to E. coli (MIC range 3.1-12.5 μM) over S. aureus (MIC range 12.5-25 μM), which follows the general observation that tobramycin is particularly active against Gram-negative organisms. Their MBC values are either equal to or slightly higher than their respective MIC values suggesting that the compounds not only are inhibiting bacterial growth but are also bactericidal. In contrast, the unconjugated peptides (P1-P4) exhibited no or little antimicrobial activity against E. coli and S. aureus. However, the MAAPCs did not show better activity than tobramycin, which is expected because our conjugates are not designed to target lab strains but instead to have high efficacy against clinically relevant pathogens such as persisters, resistant bacteria, and anaerobes that are impermeable to existing antibiotics. It should be noted that a mixture of the peptide itself with tobramycin does not enhance the antimicrobial activity compared to tobramycin alone, indicating there is no synergistic effect between the two entities when simply mixed. Whereas MAAPC05 showed similar activity against S. aureus in comparison to MAAPC01 (indicating that the terminus of conjugation did not greatly impact activity), it displayed a 2-fold increase of the MIC against E. coli. In contrast, MAAPC02, MAAPC03, and MAAPC04 displayed improved activity compared to MAAPC01. These results indicate the importance of the peptide transporter sequence as well as the conjugation site for tobramycin. Interestingly, MAAPC02 and MAAPC03 have nearly identical amino-acid composition as MAAPC01 (the only difference is a single glycine), but the amino acids are in a different sequence. In the MAAPC02 analog, the hydrophobic amino acids are clustered along one side of the -helical wheel of the peptide, which confers higher amphiphilic character and greater helical propensity. In the MAAPC03 analog, the peptide has the reversed sequence of MAAPC01. There are examples in the literature that the reversed analogs of antimicrobial peptides possess equal or enhanced antimicrobial activities. The amphiphilicity and peptide sequence orientation might play a role on the membrane-peptide interaction. However, further experiments are required to elucidate the reasons for this improved antibacterial activity.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 511145 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum bactericidal concentration (MBC) |
20.8 ± 7.2 μM
|
|||
| Administration Time | 18 h | ||||
| MOA of PDC |
We have synthesized a series of MAAPCs incorporating tobramycin with variations in the composition, sequence, and conjugation site of the peptide transporter. The MAAPCs exhibit good selectivity for bacterial cell membranes over mammalian cell membranes and do not induce any significant hemolysis of human red blood cells. MAAPCs exhibit better antibacterial activity against actively growing Gram-negative E. coli (MIC < 15 μM) than actively growing Gram-positive S. aureus (MIC < 25 μM). MAAPC01 exhibits the highest permeabilization of the outer membrane, with all other MAAPCs showing less permeation activity; tobramycin exhibits no outer membrane activity. MAAPC01 and MAAPC05 exhibit the highest inner membrane permeability, comparable to the control melittin. MAAPC02 and 03 exhibit less membrane activity, and MAAPC04 and tobramycin show negligible inner membrane activity. Higher levels of membrane activity correlate well with antimicrobial activity against persisters, where MAAPC01 and MAAPC05 show much better activity than tobramycin alone or MAAPC04.
Click to Show/Hide
|
||||
| Description |
We first investigated the antimicrobial potency of these new MAAPCs by determining their minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC) against Escherichia coli (E. coli MG1655) and Staphylococcus aureus (SA113) pathogens with a turbidity-based microdilution broth assay. Overall, all the MAAPCs display good antimicrobial activity and tend to be more selective to E. coli (MIC range 3.1-12.5 μM) over S. aureus (MIC range 12.5-25 μM), which follows the general observation that tobramycin is particularly active against Gram-negative organisms. Their MBC values are either equal to or slightly higher than their respective MIC values suggesting that the compounds not only are inhibiting bacterial growth but are also bactericidal. In contrast, the unconjugated peptides (P1-P4) exhibited no or little antimicrobial activity against E. coli and S. aureus. However, the MAAPCs did not show better activity than tobramycin, which is expected because our conjugates are not designed to target lab strains but instead to have high efficacy against clinically relevant pathogens such as persisters, resistant bacteria, and anaerobes that are impermeable to existing antibiotics. It should be noted that a mixture of the peptide itself with tobramycin does not enhance the antimicrobial activity compared to tobramycin alone, indicating there is no synergistic effect between the two entities when simply mixed. Whereas MAAPC05 showed similar activity against S. aureus in comparison to MAAPC01 (indicating that the terminus of conjugation did not greatly impact activity), it displayed a 2-fold increase of the MIC against E. coli. In contrast, MAAPC02, MAAPC03, and MAAPC04 displayed improved activity compared to MAAPC01. These results indicate the importance of the peptide transporter sequence as well as the conjugation site for tobramycin. Interestingly, MAAPC02 and MAAPC03 have nearly identical amino-acid composition as MAAPC01 (the only difference is a single glycine), but the amino acids are in a different sequence. In the MAAPC02 analog, the hydrophobic amino acids are clustered along one side of the -helical wheel of the peptide, which confers higher amphiphilic character and greater helical propensity. In the MAAPC03 analog, the peptide has the reversed sequence of MAAPC01. There are examples in the literature that the reversed analogs of antimicrobial peptides possess equal or enhanced antimicrobial activities. The amphiphilicity and peptide sequence orientation might play a role on the membrane-peptide interaction. However, further experiments are required to elucidate the reasons for this improved antibacterial activity.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 1000 μM | |||
| Administration Time | 1 h | ||||
| MOA of PDC |
We have synthesized a series of MAAPCs incorporating tobramycin with variations in the composition, sequence, and conjugation site of the peptide transporter. The MAAPCs exhibit good selectivity for bacterial cell membranes over mammalian cell membranes and do not induce any significant hemolysis of human red blood cells. MAAPCs exhibit better antibacterial activity against actively growing Gram-negative E. coli (MIC < 15 μM) than actively growing Gram-positive S. aureus (MIC < 25 μM). MAAPC01 exhibits the highest permeabilization of the outer membrane, with all other MAAPCs showing less permeation activity; tobramycin exhibits no outer membrane activity. MAAPC01 and MAAPC05 exhibit the highest inner membrane permeability, comparable to the control melittin. MAAPC02 and 03 exhibit less membrane activity, and MAAPC04 and tobramycin show negligible inner membrane activity. Higher levels of membrane activity correlate well with antimicrobial activity against persisters, where MAAPC01 and MAAPC05 show much better activity than tobramycin alone or MAAPC04.
Click to Show/Hide
|
||||
| Description |
One of the limitations to the clinical use of AMPs as antimicrobial agents is their hemolytic activity which is associated with increased toxicity. Thus, as a first assessment of the MAAPC toxicity, we investigated whether the conjugates may cause lysis of human red blood cells (hRBCs). Notably, all MAAPCs exhibited negligible hemolytic activity against hRBCs (HC50 > 500 μM), which is the first indicator of the MAAPC safety toward eukaryotic cells. Bacterial and animal cell membranes significantly differ in composition. Indeed, microbial cell surfaces contain more anionic lipids (overall negatively charged) whereas mammalian cell membranes have more lipids with neutral zwitterionic head groups (overall neutrally charged). The MAAPCs were designed to selectively discriminate between bacterial and mammalian cells. The selectivity indexes (SIs), defined as HC50/MIC, for E. coli and S. aureus demonstrated great selectivity of all MAAPCs to bacteria over hRBCs, which implies that the MAAPCs are effective against bacteria without causing harm to human cells. MAAPC04 displayed the highest bacterial selectivity (SI > 640 and SI > 160 against E. coli and S. aureus, respectively) likely due to its reduced hydrophobic content.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Human red blood cells | Homo sapiens | ||
Antimicrobial peptides (AMP) 3 - Tobramycin conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Selectivity index | > 80 | |||
| MOA of PDC |
We have synthesized a series of MAAPCs incorporating tobramycin with variations in the composition, sequence, and conjugation site of the peptide transporter. The MAAPCs exhibit good selectivity for bacterial cell membranes over mammalian cell membranes and do not induce any significant hemolysis of human red blood cells. MAAPCs exhibit better antibacterial activity against actively growing Gram-negative E. coli (MIC < 15 μM) than actively growing Gram-positive S. aureus (MIC < 25 μM). MAAPC01 exhibits the highest permeabilization of the outer membrane, with all other MAAPCs showing less permeation activity; tobramycin exhibits no outer membrane activity. MAAPC01 and MAAPC05 exhibit the highest inner membrane permeability, comparable to the control melittin. MAAPC02 and 03 exhibit less membrane activity, and MAAPC04 and tobramycin show negligible inner membrane activity. Higher levels of membrane activity correlate well with antimicrobial activity against persisters, where MAAPC01 and MAAPC05 show much better activity than tobramycin alone or MAAPC04.
Click to Show/Hide
|
||||
| Description |
One of the limitations to the clinical use of AMPs as antimicrobial agents is their hemolytic activity which is associated with increased toxicity. Thus, as a first assessment of the MAAPC toxicity, we investigated whether the conjugates may cause lysis of human red blood cells (hRBCs). Notably, all MAAPCs exhibited negligible hemolytic activity against hRBCs (HC50 > 500 μM), which is the first indicator of the MAAPC safety toward eukaryotic cells. Bacterial and animal cell membranes significantly differ in composition. Indeed, microbial cell surfaces contain more anionic lipids (overall negatively charged) whereas mammalian cell membranes have more lipids with neutral zwitterionic head groups (overall neutrally charged). The MAAPCs were designed to selectively discriminate between bacterial and mammalian cells. The selectivity indexes (SIs), defined as HC50/MIC, for E. coli and S. aureus demonstrated great selectivity of all MAAPCs to bacteria over hRBCs, which implies that the MAAPCs are effective against bacteria without causing harm to human cells. MAAPC04 displayed the highest bacterial selectivity (SI > 640 and SI > 160 against E. coli and S. aureus, respectively) likely due to its reduced hydrophobic content.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Selectivity index | > 320 | |||
| MOA of PDC |
We have synthesized a series of MAAPCs incorporating tobramycin with variations in the composition, sequence, and conjugation site of the peptide transporter. The MAAPCs exhibit good selectivity for bacterial cell membranes over mammalian cell membranes and do not induce any significant hemolysis of human red blood cells. MAAPCs exhibit better antibacterial activity against actively growing Gram-negative E. coli (MIC < 15 μM) than actively growing Gram-positive S. aureus (MIC < 25 μM). MAAPC01 exhibits the highest permeabilization of the outer membrane, with all other MAAPCs showing less permeation activity; tobramycin exhibits no outer membrane activity. MAAPC01 and MAAPC05 exhibit the highest inner membrane permeability, comparable to the control melittin. MAAPC02 and 03 exhibit less membrane activity, and MAAPC04 and tobramycin show negligible inner membrane activity. Higher levels of membrane activity correlate well with antimicrobial activity against persisters, where MAAPC01 and MAAPC05 show much better activity than tobramycin alone or MAAPC04.
Click to Show/Hide
|
||||
| Description |
One of the limitations to the clinical use of AMPs as antimicrobial agents is their hemolytic activity which is associated with increased toxicity. Thus, as a first assessment of the MAAPC toxicity, we investigated whether the conjugates may cause lysis of human red blood cells (hRBCs). Notably, all MAAPCs exhibited negligible hemolytic activity against hRBCs (HC50 > 500 μM), which is the first indicator of the MAAPC safety toward eukaryotic cells. Bacterial and animal cell membranes significantly differ in composition. Indeed, microbial cell surfaces contain more anionic lipids (overall negatively charged) whereas mammalian cell membranes have more lipids with neutral zwitterionic head groups (overall neutrally charged). The MAAPCs were designed to selectively discriminate between bacterial and mammalian cells. The selectivity indexes (SIs), defined as HC50/MIC, for E. coli and S. aureus demonstrated great selectivity of all MAAPCs to bacteria over hRBCs, which implies that the MAAPCs are effective against bacteria without causing harm to human cells. MAAPC04 displayed the highest bacterial selectivity (SI > 640 and SI > 160 against E. coli and S. aureus, respectively) likely due to its reduced hydrophobic content.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 511145 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | < 1.6 μM | |||
| Administration Time | 18 h | ||||
| MOA of PDC |
We have synthesized a series of MAAPCs incorporating tobramycin with variations in the composition, sequence, and conjugation site of the peptide transporter. The MAAPCs exhibit good selectivity for bacterial cell membranes over mammalian cell membranes and do not induce any significant hemolysis of human red blood cells. MAAPCs exhibit better antibacterial activity against actively growing Gram-negative E. coli (MIC < 15 μM) than actively growing Gram-positive S. aureus (MIC < 25 μM). MAAPC01 exhibits the highest permeabilization of the outer membrane, with all other MAAPCs showing less permeation activity; tobramycin exhibits no outer membrane activity. MAAPC01 and MAAPC05 exhibit the highest inner membrane permeability, comparable to the control melittin. MAAPC02 and 03 exhibit less membrane activity, and MAAPC04 and tobramycin show negligible inner membrane activity. Higher levels of membrane activity correlate well with antimicrobial activity against persisters, where MAAPC01 and MAAPC05 show much better activity than tobramycin alone or MAAPC04.
Click to Show/Hide
|
||||
| Description |
We first investigated the antimicrobial potency of these new MAAPCs by determining their minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC) against Escherichia coli (E. coli MG1655) and Staphylococcus aureus (SA113) pathogens with a turbidity-based microdilution broth assay. Overall, all the MAAPCs display good antimicrobial activity and tend to be more selective to E. coli (MIC range 3.1-12.5 μM) over S. aureus (MIC range 12.5-25 μM), which follows the general observation that tobramycin is particularly active against Gram-negative organisms. Their MBC values are either equal to or slightly higher than their respective MIC values suggesting that the compounds not only are inhibiting bacterial growth but are also bactericidal. In contrast, the unconjugated peptides (P1-P4) exhibited no or little antimicrobial activity against E. coli and S. aureus. However, the MAAPCs did not show better activity than tobramycin, which is expected because our conjugates are not designed to target lab strains but instead to have high efficacy against clinically relevant pathogens such as persisters, resistant bacteria, and anaerobes that are impermeable to existing antibiotics. It should be noted that a mixture of the peptide itself with tobramycin does not enhance the antimicrobial activity compared to tobramycin alone, indicating there is no synergistic effect between the two entities when simply mixed. Whereas MAAPC05 showed similar activity against S. aureus in comparison to MAAPC01 (indicating that the terminus of conjugation did not greatly impact activity), it displayed a 2-fold increase of the MIC against E. coli. In contrast, MAAPC02, MAAPC03, and MAAPC04 displayed improved activity compared to MAAPC01. These results indicate the importance of the peptide transporter sequence as well as the conjugation site for tobramycin. Interestingly, MAAPC02 and MAAPC03 have nearly identical amino-acid composition as MAAPC01 (the only difference is a single glycine), but the amino acids are in a different sequence. In the MAAPC02 analog, the hydrophobic amino acids are clustered along one side of the -helical wheel of the peptide, which confers higher amphiphilic character and greater helical propensity. In the MAAPC03 analog, the peptide has the reversed sequence of MAAPC01. There are examples in the literature that the reversed analogs of antimicrobial peptides possess equal or enhanced antimicrobial activities. The amphiphilicity and peptide sequence orientation might play a role on the membrane-peptide interaction. However, further experiments are required to elucidate the reasons for this improved antibacterial activity.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 511145 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
12.5 ± 0.0 μM
|
|||
| Administration Time | 18 h | ||||
| MOA of PDC |
We have synthesized a series of MAAPCs incorporating tobramycin with variations in the composition, sequence, and conjugation site of the peptide transporter. The MAAPCs exhibit good selectivity for bacterial cell membranes over mammalian cell membranes and do not induce any significant hemolysis of human red blood cells. MAAPCs exhibit better antibacterial activity against actively growing Gram-negative E. coli (MIC < 15 μM) than actively growing Gram-positive S. aureus (MIC < 25 μM). MAAPC01 exhibits the highest permeabilization of the outer membrane, with all other MAAPCs showing less permeation activity; tobramycin exhibits no outer membrane activity. MAAPC01 and MAAPC05 exhibit the highest inner membrane permeability, comparable to the control melittin. MAAPC02 and 03 exhibit less membrane activity, and MAAPC04 and tobramycin show negligible inner membrane activity. Higher levels of membrane activity correlate well with antimicrobial activity against persisters, where MAAPC01 and MAAPC05 show much better activity than tobramycin alone or MAAPC04.
Click to Show/Hide
|
||||
| Description |
We first investigated the antimicrobial potency of these new MAAPCs by determining their minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC) against Escherichia coli (E. coli MG1655) and Staphylococcus aureus (SA113) pathogens with a turbidity-based microdilution broth assay. Overall, all the MAAPCs display good antimicrobial activity and tend to be more selective to E. coli (MIC range 3.1-12.5 μM) over S. aureus (MIC range 12.5-25 μM), which follows the general observation that tobramycin is particularly active against Gram-negative organisms. Their MBC values are either equal to or slightly higher than their respective MIC values suggesting that the compounds not only are inhibiting bacterial growth but are also bactericidal. In contrast, the unconjugated peptides (P1-P4) exhibited no or little antimicrobial activity against E. coli and S. aureus. However, the MAAPCs did not show better activity than tobramycin, which is expected because our conjugates are not designed to target lab strains but instead to have high efficacy against clinically relevant pathogens such as persisters, resistant bacteria, and anaerobes that are impermeable to existing antibiotics. It should be noted that a mixture of the peptide itself with tobramycin does not enhance the antimicrobial activity compared to tobramycin alone, indicating there is no synergistic effect between the two entities when simply mixed. Whereas MAAPC05 showed similar activity against S. aureus in comparison to MAAPC01 (indicating that the terminus of conjugation did not greatly impact activity), it displayed a 2-fold increase of the MIC against E. coli. In contrast, MAAPC02, MAAPC03, and MAAPC04 displayed improved activity compared to MAAPC01. These results indicate the importance of the peptide transporter sequence as well as the conjugation site for tobramycin. Interestingly, MAAPC02 and MAAPC03 have nearly identical amino-acid composition as MAAPC01 (the only difference is a single glycine), but the amino acids are in a different sequence. In the MAAPC02 analog, the hydrophobic amino acids are clustered along one side of the -helical wheel of the peptide, which confers higher amphiphilic character and greater helical propensity. In the MAAPC03 analog, the peptide has the reversed sequence of MAAPC01. There are examples in the literature that the reversed analogs of antimicrobial peptides possess equal or enhanced antimicrobial activities. The amphiphilicity and peptide sequence orientation might play a role on the membrane-peptide interaction. However, further experiments are required to elucidate the reasons for this improved antibacterial activity.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum bactericidal concentration (MBC) |
3.1 ± 0.0 μM
|
|||
| Administration Time | 18 h | ||||
| MOA of PDC |
We have synthesized a series of MAAPCs incorporating tobramycin with variations in the composition, sequence, and conjugation site of the peptide transporter. The MAAPCs exhibit good selectivity for bacterial cell membranes over mammalian cell membranes and do not induce any significant hemolysis of human red blood cells. MAAPCs exhibit better antibacterial activity against actively growing Gram-negative E. coli (MIC < 15 μM) than actively growing Gram-positive S. aureus (MIC < 25 μM). MAAPC01 exhibits the highest permeabilization of the outer membrane, with all other MAAPCs showing less permeation activity; tobramycin exhibits no outer membrane activity. MAAPC01 and MAAPC05 exhibit the highest inner membrane permeability, comparable to the control melittin. MAAPC02 and 03 exhibit less membrane activity, and MAAPC04 and tobramycin show negligible inner membrane activity. Higher levels of membrane activity correlate well with antimicrobial activity against persisters, where MAAPC01 and MAAPC05 show much better activity than tobramycin alone or MAAPC04.
Click to Show/Hide
|
||||
| Description |
We first investigated the antimicrobial potency of these new MAAPCs by determining their minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC) against Escherichia coli (E. coli MG1655) and Staphylococcus aureus (SA113) pathogens with a turbidity-based microdilution broth assay. Overall, all the MAAPCs display good antimicrobial activity and tend to be more selective to E. coli (MIC range 3.1-12.5 μM) over S. aureus (MIC range 12.5-25 μM), which follows the general observation that tobramycin is particularly active against Gram-negative organisms. Their MBC values are either equal to or slightly higher than their respective MIC values suggesting that the compounds not only are inhibiting bacterial growth but are also bactericidal. In contrast, the unconjugated peptides (P1-P4) exhibited no or little antimicrobial activity against E. coli and S. aureus. However, the MAAPCs did not show better activity than tobramycin, which is expected because our conjugates are not designed to target lab strains but instead to have high efficacy against clinically relevant pathogens such as persisters, resistant bacteria, and anaerobes that are impermeable to existing antibiotics. It should be noted that a mixture of the peptide itself with tobramycin does not enhance the antimicrobial activity compared to tobramycin alone, indicating there is no synergistic effect between the two entities when simply mixed. Whereas MAAPC05 showed similar activity against S. aureus in comparison to MAAPC01 (indicating that the terminus of conjugation did not greatly impact activity), it displayed a 2-fold increase of the MIC against E. coli. In contrast, MAAPC02, MAAPC03, and MAAPC04 displayed improved activity compared to MAAPC01. These results indicate the importance of the peptide transporter sequence as well as the conjugation site for tobramycin. Interestingly, MAAPC02 and MAAPC03 have nearly identical amino-acid composition as MAAPC01 (the only difference is a single glycine), but the amino acids are in a different sequence. In the MAAPC02 analog, the hydrophobic amino acids are clustered along one side of the -helical wheel of the peptide, which confers higher amphiphilic character and greater helical propensity. In the MAAPC03 analog, the peptide has the reversed sequence of MAAPC01. There are examples in the literature that the reversed analogs of antimicrobial peptides possess equal or enhanced antimicrobial activities. The amphiphilicity and peptide sequence orientation might play a role on the membrane-peptide interaction. However, further experiments are required to elucidate the reasons for this improved antibacterial activity.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 511145 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum bactericidal concentration (MBC) |
25.0 ± 0.0 μM
|
|||
| Administration Time | 18 h | ||||
| MOA of PDC |
We have synthesized a series of MAAPCs incorporating tobramycin with variations in the composition, sequence, and conjugation site of the peptide transporter. The MAAPCs exhibit good selectivity for bacterial cell membranes over mammalian cell membranes and do not induce any significant hemolysis of human red blood cells. MAAPCs exhibit better antibacterial activity against actively growing Gram-negative E. coli (MIC < 15 μM) than actively growing Gram-positive S. aureus (MIC < 25 μM). MAAPC01 exhibits the highest permeabilization of the outer membrane, with all other MAAPCs showing less permeation activity; tobramycin exhibits no outer membrane activity. MAAPC01 and MAAPC05 exhibit the highest inner membrane permeability, comparable to the control melittin. MAAPC02 and 03 exhibit less membrane activity, and MAAPC04 and tobramycin show negligible inner membrane activity. Higher levels of membrane activity correlate well with antimicrobial activity against persisters, where MAAPC01 and MAAPC05 show much better activity than tobramycin alone or MAAPC04.
Click to Show/Hide
|
||||
| Description |
We first investigated the antimicrobial potency of these new MAAPCs by determining their minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC) against Escherichia coli (E. coli MG1655) and Staphylococcus aureus (SA113) pathogens with a turbidity-based microdilution broth assay. Overall, all the MAAPCs display good antimicrobial activity and tend to be more selective to E. coli (MIC range 3.1-12.5 μM) over S. aureus (MIC range 12.5-25 μM), which follows the general observation that tobramycin is particularly active against Gram-negative organisms. Their MBC values are either equal to or slightly higher than their respective MIC values suggesting that the compounds not only are inhibiting bacterial growth but are also bactericidal. In contrast, the unconjugated peptides (P1-P4) exhibited no or little antimicrobial activity against E. coli and S. aureus. However, the MAAPCs did not show better activity than tobramycin, which is expected because our conjugates are not designed to target lab strains but instead to have high efficacy against clinically relevant pathogens such as persisters, resistant bacteria, and anaerobes that are impermeable to existing antibiotics. It should be noted that a mixture of the peptide itself with tobramycin does not enhance the antimicrobial activity compared to tobramycin alone, indicating there is no synergistic effect between the two entities when simply mixed. Whereas MAAPC05 showed similar activity against S. aureus in comparison to MAAPC01 (indicating that the terminus of conjugation did not greatly impact activity), it displayed a 2-fold increase of the MIC against E. coli. In contrast, MAAPC02, MAAPC03, and MAAPC04 displayed improved activity compared to MAAPC01. These results indicate the importance of the peptide transporter sequence as well as the conjugation site for tobramycin. Interestingly, MAAPC02 and MAAPC03 have nearly identical amino-acid composition as MAAPC01 (the only difference is a single glycine), but the amino acids are in a different sequence. In the MAAPC02 analog, the hydrophobic amino acids are clustered along one side of the -helical wheel of the peptide, which confers higher amphiphilic character and greater helical propensity. In the MAAPC03 analog, the peptide has the reversed sequence of MAAPC01. There are examples in the literature that the reversed analogs of antimicrobial peptides possess equal or enhanced antimicrobial activities. The amphiphilicity and peptide sequence orientation might play a role on the membrane-peptide interaction. However, further experiments are required to elucidate the reasons for this improved antibacterial activity.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 1000 μM | |||
| Administration Time | 1 h | ||||
| MOA of PDC |
We have synthesized a series of MAAPCs incorporating tobramycin with variations in the composition, sequence, and conjugation site of the peptide transporter. The MAAPCs exhibit good selectivity for bacterial cell membranes over mammalian cell membranes and do not induce any significant hemolysis of human red blood cells. MAAPCs exhibit better antibacterial activity against actively growing Gram-negative E. coli (MIC < 15 μM) than actively growing Gram-positive S. aureus (MIC < 25 μM). MAAPC01 exhibits the highest permeabilization of the outer membrane, with all other MAAPCs showing less permeation activity; tobramycin exhibits no outer membrane activity. MAAPC01 and MAAPC05 exhibit the highest inner membrane permeability, comparable to the control melittin. MAAPC02 and 03 exhibit less membrane activity, and MAAPC04 and tobramycin show negligible inner membrane activity. Higher levels of membrane activity correlate well with antimicrobial activity against persisters, where MAAPC01 and MAAPC05 show much better activity than tobramycin alone or MAAPC04.
Click to Show/Hide
|
||||
| Description |
One of the limitations to the clinical use of AMPs as antimicrobial agents is their hemolytic activity which is associated with increased toxicity. Thus, as a first assessment of the MAAPC toxicity, we investigated whether the conjugates may cause lysis of human red blood cells (hRBCs). Notably, all MAAPCs exhibited negligible hemolytic activity against hRBCs (HC50 > 500 μM), which is the first indicator of the MAAPC safety toward eukaryotic cells. Bacterial and animal cell membranes significantly differ in composition. Indeed, microbial cell surfaces contain more anionic lipids (overall negatively charged) whereas mammalian cell membranes have more lipids with neutral zwitterionic head groups (overall neutrally charged). The MAAPCs were designed to selectively discriminate between bacterial and mammalian cells. The selectivity indexes (SIs), defined as HC50/MIC, for E. coli and S. aureus demonstrated great selectivity of all MAAPCs to bacteria over hRBCs, which implies that the MAAPCs are effective against bacteria without causing harm to human cells. MAAPC04 displayed the highest bacterial selectivity (SI > 640 and SI > 160 against E. coli and S. aureus, respectively) likely due to its reduced hydrophobic content.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Human red blood cells | Homo sapiens | ||
Antimicrobial peptides (AMP) 2 - Tobramycin conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Selectivity index | > 160 | |||
| MOA of PDC |
We have synthesized a series of MAAPCs incorporating tobramycin with variations in the composition, sequence, and conjugation site of the peptide transporter. The MAAPCs exhibit good selectivity for bacterial cell membranes over mammalian cell membranes and do not induce any significant hemolysis of human red blood cells. MAAPCs exhibit better antibacterial activity against actively growing Gram-negative E. coli (MIC < 15 μM) than actively growing Gram-positive S. aureus (MIC < 25 μM). MAAPC01 exhibits the highest permeabilization of the outer membrane, with all other MAAPCs showing less permeation activity; tobramycin exhibits no outer membrane activity. MAAPC01 and MAAPC05 exhibit the highest inner membrane permeability, comparable to the control melittin. MAAPC02 and 03 exhibit less membrane activity, and MAAPC04 and tobramycin show negligible inner membrane activity. Higher levels of membrane activity correlate well with antimicrobial activity against persisters, where MAAPC01 and MAAPC05 show much better activity than tobramycin alone or MAAPC04.
Click to Show/Hide
|
||||
| Description |
One of the limitations to the clinical use of AMPs as antimicrobial agents is their hemolytic activity which is associated with increased toxicity. Thus, as a first assessment of the MAAPC toxicity, we investigated whether the conjugates may cause lysis of human red blood cells (hRBCs). Notably, all MAAPCs exhibited negligible hemolytic activity against hRBCs (HC50 > 500 μM), which is the first indicator of the MAAPC safety toward eukaryotic cells. Bacterial and animal cell membranes significantly differ in composition. Indeed, microbial cell surfaces contain more anionic lipids (overall negatively charged) whereas mammalian cell membranes have more lipids with neutral zwitterionic head groups (overall neutrally charged). The MAAPCs were designed to selectively discriminate between bacterial and mammalian cells. The selectivity indexes (SIs), defined as HC50/MIC, for E. coli and S. aureus demonstrated great selectivity of all MAAPCs to bacteria over hRBCs, which implies that the MAAPCs are effective against bacteria without causing harm to human cells. MAAPC04 displayed the highest bacterial selectivity (SI > 640 and SI > 160 against E. coli and S. aureus, respectively) likely due to its reduced hydrophobic content.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Selectivity index | > 640 | |||
| MOA of PDC |
We have synthesized a series of MAAPCs incorporating tobramycin with variations in the composition, sequence, and conjugation site of the peptide transporter. The MAAPCs exhibit good selectivity for bacterial cell membranes over mammalian cell membranes and do not induce any significant hemolysis of human red blood cells. MAAPCs exhibit better antibacterial activity against actively growing Gram-negative E. coli (MIC < 15 μM) than actively growing Gram-positive S. aureus (MIC < 25 μM). MAAPC01 exhibits the highest permeabilization of the outer membrane, with all other MAAPCs showing less permeation activity; tobramycin exhibits no outer membrane activity. MAAPC01 and MAAPC05 exhibit the highest inner membrane permeability, comparable to the control melittin. MAAPC02 and 03 exhibit less membrane activity, and MAAPC04 and tobramycin show negligible inner membrane activity. Higher levels of membrane activity correlate well with antimicrobial activity against persisters, where MAAPC01 and MAAPC05 show much better activity than tobramycin alone or MAAPC04.
Click to Show/Hide
|
||||
| Description |
One of the limitations to the clinical use of AMPs as antimicrobial agents is their hemolytic activity which is associated with increased toxicity. Thus, as a first assessment of the MAAPC toxicity, we investigated whether the conjugates may cause lysis of human red blood cells (hRBCs). Notably, all MAAPCs exhibited negligible hemolytic activity against hRBCs (HC50 > 500 μM), which is the first indicator of the MAAPC safety toward eukaryotic cells. Bacterial and animal cell membranes significantly differ in composition. Indeed, microbial cell surfaces contain more anionic lipids (overall negatively charged) whereas mammalian cell membranes have more lipids with neutral zwitterionic head groups (overall neutrally charged). The MAAPCs were designed to selectively discriminate between bacterial and mammalian cells. The selectivity indexes (SIs), defined as HC50/MIC, for E. coli and S. aureus demonstrated great selectivity of all MAAPCs to bacteria over hRBCs, which implies that the MAAPCs are effective against bacteria without causing harm to human cells. MAAPC04 displayed the highest bacterial selectivity (SI > 640 and SI > 160 against E. coli and S. aureus, respectively) likely due to its reduced hydrophobic content.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 511145 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
3.1 ± 0.0 μM
|
|||
| Administration Time | 18 h | ||||
| MOA of PDC |
We have synthesized a series of MAAPCs incorporating tobramycin with variations in the composition, sequence, and conjugation site of the peptide transporter. The MAAPCs exhibit good selectivity for bacterial cell membranes over mammalian cell membranes and do not induce any significant hemolysis of human red blood cells. MAAPCs exhibit better antibacterial activity against actively growing Gram-negative E. coli (MIC < 15 μM) than actively growing Gram-positive S. aureus (MIC < 25 μM). MAAPC01 exhibits the highest permeabilization of the outer membrane, with all other MAAPCs showing less permeation activity; tobramycin exhibits no outer membrane activity. MAAPC01 and MAAPC05 exhibit the highest inner membrane permeability, comparable to the control melittin. MAAPC02 and 03 exhibit less membrane activity, and MAAPC04 and tobramycin show negligible inner membrane activity. Higher levels of membrane activity correlate well with antimicrobial activity against persisters, where MAAPC01 and MAAPC05 show much better activity than tobramycin alone or MAAPC04.
Click to Show/Hide
|
||||
| Description |
We first investigated the antimicrobial potency of these new MAAPCs by determining their minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC) against Escherichia coli (E. coli MG1655) and Staphylococcus aureus (SA113) pathogens with a turbidity-based microdilution broth assay. Overall, all the MAAPCs display good antimicrobial activity and tend to be more selective to E. coli (MIC range 3.1-12.5 μM) over S. aureus (MIC range 12.5-25 μM), which follows the general observation that tobramycin is particularly active against Gram-negative organisms. Their MBC values are either equal to or slightly higher than their respective MIC values suggesting that the compounds not only are inhibiting bacterial growth but are also bactericidal. In contrast, the unconjugated peptides (P1-P4) exhibited no or little antimicrobial activity against E. coli and S. aureus. However, the MAAPCs did not show better activity than tobramycin, which is expected because our conjugates are not designed to target lab strains but instead to have high efficacy against clinically relevant pathogens such as persisters, resistant bacteria, and anaerobes that are impermeable to existing antibiotics. It should be noted that a mixture of the peptide itself with tobramycin does not enhance the antimicrobial activity compared to tobramycin alone, indicating there is no synergistic effect between the two entities when simply mixed. Whereas MAAPC05 showed similar activity against S. aureus in comparison to MAAPC01 (indicating that the terminus of conjugation did not greatly impact activity), it displayed a 2-fold increase of the MIC against E. coli. In contrast, MAAPC02, MAAPC03, and MAAPC04 displayed improved activity compared to MAAPC01. These results indicate the importance of the peptide transporter sequence as well as the conjugation site for tobramycin. Interestingly, MAAPC02 and MAAPC03 have nearly identical amino-acid composition as MAAPC01 (the only difference is a single glycine), but the amino acids are in a different sequence. In the MAAPC02 analog, the hydrophobic amino acids are clustered along one side of the -helical wheel of the peptide, which confers higher amphiphilic character and greater helical propensity. In the MAAPC03 analog, the peptide has the reversed sequence of MAAPC01. There are examples in the literature that the reversed analogs of antimicrobial peptides possess equal or enhanced antimicrobial activities. The amphiphilicity and peptide sequence orientation might play a role on the membrane-peptide interaction. However, further experiments are required to elucidate the reasons for this improved antibacterial activity.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 511145 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
12.5 ± 0.0 μM
|
|||
| Administration Time | 18 h | ||||
| MOA of PDC |
We have synthesized a series of MAAPCs incorporating tobramycin with variations in the composition, sequence, and conjugation site of the peptide transporter. The MAAPCs exhibit good selectivity for bacterial cell membranes over mammalian cell membranes and do not induce any significant hemolysis of human red blood cells. MAAPCs exhibit better antibacterial activity against actively growing Gram-negative E. coli (MIC < 15 μM) than actively growing Gram-positive S. aureus (MIC < 25 μM). MAAPC01 exhibits the highest permeabilization of the outer membrane, with all other MAAPCs showing less permeation activity; tobramycin exhibits no outer membrane activity. MAAPC01 and MAAPC05 exhibit the highest inner membrane permeability, comparable to the control melittin. MAAPC02 and 03 exhibit less membrane activity, and MAAPC04 and tobramycin show negligible inner membrane activity. Higher levels of membrane activity correlate well with antimicrobial activity against persisters, where MAAPC01 and MAAPC05 show much better activity than tobramycin alone or MAAPC04.
Click to Show/Hide
|
||||
| Description |
We first investigated the antimicrobial potency of these new MAAPCs by determining their minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC) against Escherichia coli (E. coli MG1655) and Staphylococcus aureus (SA113) pathogens with a turbidity-based microdilution broth assay. Overall, all the MAAPCs display good antimicrobial activity and tend to be more selective to E. coli (MIC range 3.1-12.5 μM) over S. aureus (MIC range 12.5-25 μM), which follows the general observation that tobramycin is particularly active against Gram-negative organisms. Their MBC values are either equal to or slightly higher than their respective MIC values suggesting that the compounds not only are inhibiting bacterial growth but are also bactericidal. In contrast, the unconjugated peptides (P1-P4) exhibited no or little antimicrobial activity against E. coli and S. aureus. However, the MAAPCs did not show better activity than tobramycin, which is expected because our conjugates are not designed to target lab strains but instead to have high efficacy against clinically relevant pathogens such as persisters, resistant bacteria, and anaerobes that are impermeable to existing antibiotics. It should be noted that a mixture of the peptide itself with tobramycin does not enhance the antimicrobial activity compared to tobramycin alone, indicating there is no synergistic effect between the two entities when simply mixed. Whereas MAAPC05 showed similar activity against S. aureus in comparison to MAAPC01 (indicating that the terminus of conjugation did not greatly impact activity), it displayed a 2-fold increase of the MIC against E. coli. In contrast, MAAPC02, MAAPC03, and MAAPC04 displayed improved activity compared to MAAPC01. These results indicate the importance of the peptide transporter sequence as well as the conjugation site for tobramycin. Interestingly, MAAPC02 and MAAPC03 have nearly identical amino-acid composition as MAAPC01 (the only difference is a single glycine), but the amino acids are in a different sequence. In the MAAPC02 analog, the hydrophobic amino acids are clustered along one side of the -helical wheel of the peptide, which confers higher amphiphilic character and greater helical propensity. In the MAAPC03 analog, the peptide has the reversed sequence of MAAPC01. There are examples in the literature that the reversed analogs of antimicrobial peptides possess equal or enhanced antimicrobial activities. The amphiphilicity and peptide sequence orientation might play a role on the membrane-peptide interaction. However, further experiments are required to elucidate the reasons for this improved antibacterial activity.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum bactericidal concentration (MBC) |
4.2 ± 1.8 μM
|
|||
| Administration Time | 18 h | ||||
| MOA of PDC |
We have synthesized a series of MAAPCs incorporating tobramycin with variations in the composition, sequence, and conjugation site of the peptide transporter. The MAAPCs exhibit good selectivity for bacterial cell membranes over mammalian cell membranes and do not induce any significant hemolysis of human red blood cells. MAAPCs exhibit better antibacterial activity against actively growing Gram-negative E. coli (MIC < 15 μM) than actively growing Gram-positive S. aureus (MIC < 25 μM). MAAPC01 exhibits the highest permeabilization of the outer membrane, with all other MAAPCs showing less permeation activity; tobramycin exhibits no outer membrane activity. MAAPC01 and MAAPC05 exhibit the highest inner membrane permeability, comparable to the control melittin. MAAPC02 and 03 exhibit less membrane activity, and MAAPC04 and tobramycin show negligible inner membrane activity. Higher levels of membrane activity correlate well with antimicrobial activity against persisters, where MAAPC01 and MAAPC05 show much better activity than tobramycin alone or MAAPC04.
Click to Show/Hide
|
||||
| Description |
We first investigated the antimicrobial potency of these new MAAPCs by determining their minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC) against Escherichia coli (E. coli MG1655) and Staphylococcus aureus (SA113) pathogens with a turbidity-based microdilution broth assay. Overall, all the MAAPCs display good antimicrobial activity and tend to be more selective to E. coli (MIC range 3.1-12.5 μM) over S. aureus (MIC range 12.5-25 μM), which follows the general observation that tobramycin is particularly active against Gram-negative organisms. Their MBC values are either equal to or slightly higher than their respective MIC values suggesting that the compounds not only are inhibiting bacterial growth but are also bactericidal. In contrast, the unconjugated peptides (P1-P4) exhibited no or little antimicrobial activity against E. coli and S. aureus. However, the MAAPCs did not show better activity than tobramycin, which is expected because our conjugates are not designed to target lab strains but instead to have high efficacy against clinically relevant pathogens such as persisters, resistant bacteria, and anaerobes that are impermeable to existing antibiotics. It should be noted that a mixture of the peptide itself with tobramycin does not enhance the antimicrobial activity compared to tobramycin alone, indicating there is no synergistic effect between the two entities when simply mixed. Whereas MAAPC05 showed similar activity against S. aureus in comparison to MAAPC01 (indicating that the terminus of conjugation did not greatly impact activity), it displayed a 2-fold increase of the MIC against E. coli. In contrast, MAAPC02, MAAPC03, and MAAPC04 displayed improved activity compared to MAAPC01. These results indicate the importance of the peptide transporter sequence as well as the conjugation site for tobramycin. Interestingly, MAAPC02 and MAAPC03 have nearly identical amino-acid composition as MAAPC01 (the only difference is a single glycine), but the amino acids are in a different sequence. In the MAAPC02 analog, the hydrophobic amino acids are clustered along one side of the -helical wheel of the peptide, which confers higher amphiphilic character and greater helical propensity. In the MAAPC03 analog, the peptide has the reversed sequence of MAAPC01. There are examples in the literature that the reversed analogs of antimicrobial peptides possess equal or enhanced antimicrobial activities. The amphiphilicity and peptide sequence orientation might play a role on the membrane-peptide interaction. However, further experiments are required to elucidate the reasons for this improved antibacterial activity.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 511145 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum bactericidal concentration (MBC) |
14.6 ± 9.5 μM
|
|||
| Administration Time | 18 h | ||||
| MOA of PDC |
We have synthesized a series of MAAPCs incorporating tobramycin with variations in the composition, sequence, and conjugation site of the peptide transporter. The MAAPCs exhibit good selectivity for bacterial cell membranes over mammalian cell membranes and do not induce any significant hemolysis of human red blood cells. MAAPCs exhibit better antibacterial activity against actively growing Gram-negative E. coli (MIC < 15 μM) than actively growing Gram-positive S. aureus (MIC < 25 μM). MAAPC01 exhibits the highest permeabilization of the outer membrane, with all other MAAPCs showing less permeation activity; tobramycin exhibits no outer membrane activity. MAAPC01 and MAAPC05 exhibit the highest inner membrane permeability, comparable to the control melittin. MAAPC02 and 03 exhibit less membrane activity, and MAAPC04 and tobramycin show negligible inner membrane activity. Higher levels of membrane activity correlate well with antimicrobial activity against persisters, where MAAPC01 and MAAPC05 show much better activity than tobramycin alone or MAAPC04.
Click to Show/Hide
|
||||
| Description |
We first investigated the antimicrobial potency of these new MAAPCs by determining their minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC) against Escherichia coli (E. coli MG1655) and Staphylococcus aureus (SA113) pathogens with a turbidity-based microdilution broth assay. Overall, all the MAAPCs display good antimicrobial activity and tend to be more selective to E. coli (MIC range 3.1-12.5 μM) over S. aureus (MIC range 12.5-25 μM), which follows the general observation that tobramycin is particularly active against Gram-negative organisms. Their MBC values are either equal to or slightly higher than their respective MIC values suggesting that the compounds not only are inhibiting bacterial growth but are also bactericidal. In contrast, the unconjugated peptides (P1-P4) exhibited no or little antimicrobial activity against E. coli and S. aureus. However, the MAAPCs did not show better activity than tobramycin, which is expected because our conjugates are not designed to target lab strains but instead to have high efficacy against clinically relevant pathogens such as persisters, resistant bacteria, and anaerobes that are impermeable to existing antibiotics. It should be noted that a mixture of the peptide itself with tobramycin does not enhance the antimicrobial activity compared to tobramycin alone, indicating there is no synergistic effect between the two entities when simply mixed. Whereas MAAPC05 showed similar activity against S. aureus in comparison to MAAPC01 (indicating that the terminus of conjugation did not greatly impact activity), it displayed a 2-fold increase of the MIC against E. coli. In contrast, MAAPC02, MAAPC03, and MAAPC04 displayed improved activity compared to MAAPC01. These results indicate the importance of the peptide transporter sequence as well as the conjugation site for tobramycin. Interestingly, MAAPC02 and MAAPC03 have nearly identical amino-acid composition as MAAPC01 (the only difference is a single glycine), but the amino acids are in a different sequence. In the MAAPC02 analog, the hydrophobic amino acids are clustered along one side of the -helical wheel of the peptide, which confers higher amphiphilic character and greater helical propensity. In the MAAPC03 analog, the peptide has the reversed sequence of MAAPC01. There are examples in the literature that the reversed analogs of antimicrobial peptides possess equal or enhanced antimicrobial activities. The amphiphilicity and peptide sequence orientation might play a role on the membrane-peptide interaction. However, further experiments are required to elucidate the reasons for this improved antibacterial activity.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 2000 μM | |||
| Administration Time | 1 h | ||||
| MOA of PDC |
We have synthesized a series of MAAPCs incorporating tobramycin with variations in the composition, sequence, and conjugation site of the peptide transporter. The MAAPCs exhibit good selectivity for bacterial cell membranes over mammalian cell membranes and do not induce any significant hemolysis of human red blood cells. MAAPCs exhibit better antibacterial activity against actively growing Gram-negative E. coli (MIC < 15 μM) than actively growing Gram-positive S. aureus (MIC < 25 μM). MAAPC01 exhibits the highest permeabilization of the outer membrane, with all other MAAPCs showing less permeation activity; tobramycin exhibits no outer membrane activity. MAAPC01 and MAAPC05 exhibit the highest inner membrane permeability, comparable to the control melittin. MAAPC02 and 03 exhibit less membrane activity, and MAAPC04 and tobramycin show negligible inner membrane activity. Higher levels of membrane activity correlate well with antimicrobial activity against persisters, where MAAPC01 and MAAPC05 show much better activity than tobramycin alone or MAAPC04.
Click to Show/Hide
|
||||
| Description |
One of the limitations to the clinical use of AMPs as antimicrobial agents is their hemolytic activity which is associated with increased toxicity. Thus, as a first assessment of the MAAPC toxicity, we investigated whether the conjugates may cause lysis of human red blood cells (hRBCs). Notably, all MAAPCs exhibited negligible hemolytic activity against hRBCs (HC50 > 500 μM), which is the first indicator of the MAAPC safety toward eukaryotic cells. Bacterial and animal cell membranes significantly differ in composition. Indeed, microbial cell surfaces contain more anionic lipids (overall negatively charged) whereas mammalian cell membranes have more lipids with neutral zwitterionic head groups (overall neutrally charged). The MAAPCs were designed to selectively discriminate between bacterial and mammalian cells. The selectivity indexes (SIs), defined as HC50/MIC, for E. coli and S. aureus demonstrated great selectivity of all MAAPCs to bacteria over hRBCs, which implies that the MAAPCs are effective against bacteria without causing harm to human cells. MAAPC04 displayed the highest bacterial selectivity (SI > 640 and SI > 160 against E. coli and S. aureus, respectively) likely due to its reduced hydrophobic content.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Human red blood cells | Homo sapiens | ||
OGF-Gem [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [14] | ||||
| Indication | Pancreatic ductal adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
17.63 ± 2.334 nM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Therefore, we designed, synthesized, and characterized an OGF-Gem conjugate, where OGF and Gem are tethered by an organic linker (Figure 1). Gem was subjected to selective protection using the tert-butoxycarbonyl (Boc) group and prepared as gemcitabine hemisuccinate. 5-O-diBoc-gemcitabine hemisuccinate was conjugated with the OGF peptide in solution. We demonstrated the cytotoxic activity of the OGF-Gem conjugate against pancreatic cancer cell lines, including the metastatic line (MIA PaCa-2 and AsPC-1). Furthermore, we confirmed that OGF-Gem is either not cytotoxic or significantly less cytotoxic to two non-tumor-transformed human cellskidney (HEK-293) and skin fibroblast cells (HDFa). We also determined the effect of OGF-Gem on cell cycle inhibition, and the inhibition of cell proliferation, senescent cells, and apoptosis. We have demonstrated that OGF-Gem has antimetastatic potential due to inhibited pancreatic tumor cell (AsPC-1)-induced platelet aggregation. This can significantly impact the inhibition of disease progression (metastasis) of pancreatic cancer.
Click to Show/Hide
|
||||
| Description |
The tested compounds cytotoxic activity was determined using the MTT test, which is based on the ability to convert tetrazole salts to water insoluble formazan through mitochondrial dehydrogenases. Our results show a high cytotoxic effect on all pancreatic cancer cell lines. Exposing pancreatic cell lines MIA PaCa-2 and AsPC-1 to OGF-Gem decreased viability. Importantly, the OGF-Gem conjugate demonstrated a more pronounced cytotoxic effect against the metastatic pancreatic cancer cell line AsPC-1 compared to the commonly used chemotherapeutic agent. The results obtained for non-tumor-transformed cellsa human embryonic kidney line HEK-293 and human primary dermal fibroblast line HDFa presented a slight cytotoxicity effect from the OGF-Gem derivative within 3 days of incubation for all tested concentrations. Interestingly, an 80% reduction in HEK-293 cell viability was observed for the 100 nM Gem compared to the 100 nM OGF-Gem derivative. In HDFa cells, 100 nM Gem reduced viability to 35%, while the OGF-Gem conjugate slightly decreased the viability (to 75% viability) after 72 h of incubation. Based on the analysis of the results, concentrations of 3.125, 12.5, 50, and 100 nM, as well as an incubation time of 72 h, were selected for further experiments on the three pancreatic cancer cell lines.
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | MIA PaCa-2 cell | CVCL_0428 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [14] | ||||
| Indication | Pancreatic ductal adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
27.44 ± 9.161 nM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Therefore, we designed, synthesized, and characterized an OGF-Gem conjugate, where OGF and Gem are tethered by an organic linker (Figure 1). Gem was subjected to selective protection using the tert-butoxycarbonyl (Boc) group and prepared as gemcitabine hemisuccinate. 5-O-diBoc-gemcitabine hemisuccinate was conjugated with the OGF peptide in solution. We demonstrated the cytotoxic activity of the OGF-Gem conjugate against pancreatic cancer cell lines, including the metastatic line (MIA PaCa-2 and AsPC-1). Furthermore, we confirmed that OGF-Gem is either not cytotoxic or significantly less cytotoxic to two non-tumor-transformed human cellskidney (HEK-293) and skin fibroblast cells (HDFa). We also determined the effect of OGF-Gem on cell cycle inhibition, and the inhibition of cell proliferation, senescent cells, and apoptosis. We have demonstrated that OGF-Gem has antimetastatic potential due to inhibited pancreatic tumor cell (AsPC-1)-induced platelet aggregation. This can significantly impact the inhibition of disease progression (metastasis) of pancreatic cancer.
Click to Show/Hide
|
||||
| Description |
The tested compounds cytotoxic activity was determined using the MTT test, which is based on the ability to convert tetrazole salts to water insoluble formazan through mitochondrial dehydrogenases. Our results show a high cytotoxic effect on all pancreatic cancer cell lines. Exposing pancreatic cell lines MIA PaCa-2 and AsPC-1 to OGF-Gem decreased viability. Importantly, the OGF-Gem conjugate demonstrated a more pronounced cytotoxic effect against the metastatic pancreatic cancer cell line AsPC-1 compared to the commonly used chemotherapeutic agent. The results obtained for non-tumor-transformed cellsa human embryonic kidney line HEK-293 and human primary dermal fibroblast line HDFa presented a slight cytotoxicity effect from the OGF-Gem derivative within 3 days of incubation for all tested concentrations. Interestingly, an 80% reduction in HEK-293 cell viability was observed for the 100 nM Gem compared to the 100 nM OGF-Gem derivative. In HDFa cells, 100 nM Gem reduced viability to 35%, while the OGF-Gem conjugate slightly decreased the viability (to 75% viability) after 72 h of incubation. Based on the analysis of the results, concentrations of 3.125, 12.5, 50, and 100 nM, as well as an incubation time of 72 h, were selected for further experiments on the three pancreatic cancer cell lines.
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | AsPC-1 cell | CVCL_0152 | ||
CPP-SA-PTX [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Malignant glioblastoma | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) |
20.55 ± 1.02 nM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
A highly sensitive, nontoxic, hydrophilic cell-penetrating peptide (CPP = c[RGDKLAK]) was selected for the construction of an effective peptide-drug conjugate (PDC). A hydrophobic drug paclitaxel (PTX) was successfully conjugated with CPP via ester linkage with succinic acid (SA) as a pH-cleavable linker moiety. The characterization techniques employed in this study indicate the >95% purity of the resulting PDC (CPP-SA-PTX). The in vitro studies show that our proposed PDC exhibits enhanced stability (˜90%) and cytotoxicity (EC50 = 8.32 ± 0.09 nM). Besides the excellent solubility of PDC in water, the PTX effect on positive β-tubulin-III indicates that the drug releases retained pharmacological properties. Additionally, in vivo, therapeutic-dose treatment reveals the prominent tumor-growth inhibitory effects (2.82-3.24-fold) of PDC in tumor mice models. Subsequently, these observations confirmed that our novel-designed PDC (CPP-SA-PTX) adduct may serve as a promising therapeutic agent to treat glioblastoma.
Click to Show/Hide
|
||||
| Description |
Furthermore, it was also observed that PDC shows time-dependent cytotoxicity against U87MG cells, as at 12 h drug treatment. PDC shows a slightly lower inhibitory effect on cell survival (EC50 = 25.82 ± 0.31 nM) than PTX alone (EC50 = 12.25 ± 0.13 nM), and CPP-SA alone shows higher EC50 = 54.37 ± 0.24 in U87MG cells. Besides this, it was previously observed that CPP-treated healthy VERO cells showed 88.05 ± 0.86% viable cells even using 10 uM concentration, indicating the specificity of CPP for glioblastoma cells. However, at 24 h incubation period, our PDC shows a significantly enhanced cell survival inhibitory effect with EC50 = 8.32 ± 0.09 nM, suggesting the delayed effect of PDC because of the time required for the intracellular pH to cause maximal cleavage of PTX from the PDC. Moreover, it also shows that CPP-SA has a lower cytotoxicity effect on glioblastoma cells compared with PTX alone and PDC, indicating its specificity as a carrier and selectivity for integrin receptors overexpressed on the cell surface. Additionally, we have also studied the potential of our novel-designed PDC to internalize into PTX-resistant glioblastoma (U87MG-PR) cells to show cytotoxic activity upon intracellular cleavage of PTX from CPP. The data presented in Figure 3b reveal approximately 15% viability increases in U87MG-PR cells compared with parent U87MG cells. It can be seen in the inset in Figure 3b that PTX alone (EC50 = 41.3 ± 1.5 nM) showed significantly 2-fold reduced cytotoxic activity in U87MG-PR cells in comparison with our novel-designed PDC having EC50 = 20.55 ± 1.02 nM. It was also observed in Figure 3b that at lower concentrations (0-10 nM) the viable cell count was ˜75%; however, it significantly decreased to ˜20% viability with an increase in concentration (40 nM) of the test samples, highlighting the dose-dependent cytotoxicity behavior of PDC in a 24 h incubation period.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U87MG-PR cell | CVCL_0022 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Malignant glioblastoma | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) |
25.82 ± 0.31 nM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
A highly sensitive, nontoxic, hydrophilic cell-penetrating peptide (CPP = c[RGDKLAK]) was selected for the construction of an effective peptide-drug conjugate (PDC). A hydrophobic drug paclitaxel (PTX) was successfully conjugated with CPP via ester linkage with succinic acid (SA) as a pH-cleavable linker moiety. The characterization techniques employed in this study indicate the >95% purity of the resulting PDC (CPP-SA-PTX). The in vitro studies show that our proposed PDC exhibits enhanced stability (˜90%) and cytotoxicity (EC50 = 8.32 ± 0.09 nM). Besides the excellent solubility of PDC in water, the PTX effect on positive β-tubulin-III indicates that the drug releases retained pharmacological properties. Additionally, in vivo, therapeutic-dose treatment reveals the prominent tumor-growth inhibitory effects (2.82-3.24-fold) of PDC in tumor mice models. Subsequently, these observations confirmed that our novel-designed PDC (CPP-SA-PTX) adduct may serve as a promising therapeutic agent to treat glioblastoma.
Click to Show/Hide
|
||||
| Description |
Furthermore, it was also observed that PDC shows time-dependent cytotoxicity against U87MG cells, as at 12 h drug treatment. PDC shows a slightly lower inhibitory effect on cell survival (EC50 = 25.82 ± 0.31 nM) than PTX alone (EC50 = 12.25 ± 0.13 nM), and CPP-SA alone shows higher EC50 = 54.37 ± 0.24 in U87MG cells. Besides this, it was previously observed that CPP-treated healthy VERO cells showed 88.05 ± 0.86% viable cells even using 10 uM concentration, indicating the specificity of CPP for glioblastoma cells. However, at 24 h incubation period, our PDC shows a significantly enhanced cell survival inhibitory effect with EC50 = 8.32 ± 0.09 nM, suggesting the delayed effect of PDC because of the time required for the intracellular pH to cause maximal cleavage of PTX from the PDC. Moreover, it also shows that CPP-SA has a lower cytotoxicity effect on glioblastoma cells compared with PTX alone and PDC, indicating its specificity as a carrier and selectivity for integrin receptors overexpressed on the cell surface. Additionally, we have also studied the potential of our novel-designed PDC to internalize into PTX-resistant glioblastoma (U87MG-PR) cells to show cytotoxic activity upon intracellular cleavage of PTX from CPP. The data presented in Figure 3b reveal approximately 15% viability increases in U87MG-PR cells compared with parent U87MG cells. It can be seen in the inset in Figure 3b that PTX alone (EC50 = 41.3 ± 1.5 nM) showed significantly 2-fold reduced cytotoxic activity in U87MG-PR cells in comparison with our novel-designed PDC having EC50 = 20.55 ± 1.02 nM. It was also observed in Figure 3b that at lower concentrations (0-10 nM) the viable cell count was ˜75%; however, it significantly decreased to ˜20% viability with an increase in concentration (40 nM) of the test samples, highlighting the dose-dependent cytotoxicity behavior of PDC in a 24 h incubation period.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U-87MG cell | CVCL_0022 | ||
References